Duosol Mit 4 Mmol/L Kalium Hämofiltrationslösung
FACHINFORMATION (ZUSAMMENFASSUNG DER MERKMALE DES ARZNEIMITTELS)
1. BEZEICHNUNG DES ARZNEIMITTELS
Duosol mit 4 mmol/l Kalium Hämofiltrationslösung
2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
|
Wirkstoffe: |
Kleine Kammer Elektrolytlösung |
Große Kammer Bicarbonatlösung | ||
|
555 ml enthalten |
pro 1000 ml |
4445 ml enthalten |
pro 1000 ml | |
|
Natriumchlorid |
2,34 g |
4,21 g |
27,47 g |
6,18 g |
|
Kaliumchlorid |
1,49 g |
2,68 g |
— |
— |
|
Calciumchlorid-Dihydrat |
1,10 g |
1,98 g |
— |
— |
|
Magne siumchlorid-Hexahydrat |
0,51 g |
0,91 g |
— |
— |
|
Glucose-Monohydrat entsprechend Glucose, wasserfrei |
5,49 g 5,0 g |
9,90 g 9,0 g |
— |
— |
|
Natriumhydrogencarbonat |
— |
— |
15,96 g |
3,59 g |
|
Elektrolyte: |
[mmol/ Kammer] |
[mmol/l] |
[mmol/ Kammer] |
[mmol/l] |
|
Na+ |
40,0 |
72 |
660 |
149 |
|
K+ |
20,0 |
36,0 |
— |
— |
|
Ca2+ |
7,5 |
13,5 |
— |
— |
|
Mg2+ |
2,5 |
4,5 |
— |
— |
|
ci- |
95,0 |
171 |
470 |
106 |
|
HCO3- |
— |
— |
190 |
42,8 |
|
Theoretische Osmolarität [mOsm/l] |
34 |
17 |
297 | |
Zusammensetzung der gebrauchsfertigen Hämofiltrationslösung nach dem Mischen:
1000 ml gebrauchsfertige Hämofiltrationslösung enthalten [mmol/l]:
HCO3- 35,0
Glucose, wasserfrei 5,6 (entspr. 1,0 g)
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
3. DARREICHUNGSFORM
Hämofiltrationslösung
Klare und farblose Lösung, frei von sichtbaren Partikeln
Theoretische Osmolarität: 300 mOsm/l pH: 7,0-8,0
4. KLINISCHE ANGABEN
4.1 Anwendungsgebiete
Die gebrauchsfertige Lösung wird angewendet bei Patienten mit akutem Nierenversagen jeglicher Genese, bei denen eine kontinuierliche Hämofiltration erforderlich ist.
4.2 Dosierung und Art der Anwendung
Die Anwendung von Hämofiltrationslösungen bei Patienten mit akutem Nierenversagen sollte unter Aufsicht eines Arztes, der Erfahrung in der Anwendung solcher Behandlungen hat, erfolgen.
Dosierung
Die verordnete Filtrationsrate hängt vom klinischen Zustand und Körpergewicht des Patienten ab. Abhängig vom metabolischen Zustand des Patienten wird, soweit nicht anders verordnet, eine Filtrationsrate von 20-25 ml/kg Körpergewicht pro Stunde zur Entfernung metabolischer Abbauprodukte, die normalerweise mit dem Urin ausgeschieden werden, empfohlen.
Das Dosisvolumen liegt im Ermessen des Arztes, da das Volumen der Substitutionslösung abhängig ist von der Intensität der durchgeführten Behandlung sowie von der zu ersetzenden Flüssigkeitsmenge, die zum Erreichen eines Flüssigkeits-Gleichgewichts erforderlich ist.
Kinder und Jugendliche
Die oben angegebenen Dosierungsempfehlungen gelten auch für Kinder und Jugendliche.
Art der Anwendung
Intravenöse Anwendung.
Die gebrauchsfertige Hämofiltrationslösung muss durch Öffnen der Peelnaht hergestellt werden. Durch fünfmaliges Wenden des Beutels wird die Mischung erhalten. Weitere Hinweise siehe Abschnitt 6.6.
Die gebrauchsfertige Hämofiltrationslösung wird mittels einer Dosierpumpeneinheit in den extrakorporalen Kreislauf infundiert.
Die Hämofiltrationslösung ersetzt das bei der Hämofiltration dem Blut entzogene Ultrafiltrat unter Berücksichtigung der Gesamtflüssigkeitsbilanz.
Die Behandlung bei akutem Nierenversagen ist zeitlich begrenzt und wird bei vollständiger Wiederherstellung der Nierenfunktion beendet.
4.3 Gegenanzeigen
Spezifische Gegenanzeigen aufgrund der gebrauchsfertigen Hämofiltrationslösung:
• Hyperkaliämie
• Metabolische Alkalose
Allgemeine Gegenanzeigen aufgrund der Hämofiltration:
• Akutes Nierenversagen mit ausgeprägtem Hyperkatabolismus, wenn die urämische Symptomatik mit der Hämofiltration nicht mehr behoben werden kann
• Unzureichender Blutfluss aus dem Blutgefäßzugang
• Alle Zustände mit erhöhtem Blutungsrisiko aufgrund systemischer Antikoagulation
4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Vor und während der Hämofiltration sollten der hämodynamische Zustand sowie das Flüssigkeits-, Elektrolyt- und Säure-Basen-Gleichgewicht, die Blutglucosekonzentration und die Harnstoff- und Plasmakreatininspiegel engmaschig überwacht werden.
Die Kaliumkonzentration im Serum muss vor und während der Hämofiltration regelmäßig kontrolliert werden. Bei Abnahme der Kaliumkonzentration im Serum und Entstehung einer Hypokaliämie kann eine Kaliumsubstitution erforderlich werden. Bei erhöhter Kaliumkonzentration im Serum, einer Hyperkaliämie, kann es notwendig sein, neben den sonstigen Maßnahmen der Intensivmedizin einen Anstieg der Filtrationsrate und/oder einen Wechsel zu einer Substitutionslösung mit einer geringeren Kaliumkonzentration in Betracht zu ziehen.
Während der Hämofiltration sollte die anorganische Phosphatkonzentration regelmäßig gemessen werden.
Im Falle einer Hypophosphatämie müssen anorganische Phosphate substituiert werden.
Behältnisse aus Kunststoff können gelegentlich während des Transports vom Hersteller in das Krankenhaus/Dialysezentrum oder innerhalb des Krankenhauses/Dialysezentrums beschädigt werden. Dadurch kann es zu einer Kontamination mit mikrobiellem oder fungiformen Wachstum in der Hämofiltrationslösung kommen. Eine sorgfältige Sichtkontrolle des Behältnisses und der Hämofiltrationslösung ist daher vor dem Anschließen des Behältnisses und vor Anwendung der Lösung erforderlich. Dabei ist insbesondere auf geringste Beschädigungen am Verschluss, an den Verschlussnähten, an der Peelnaht sowie an den Behältnisecken als Quelle einer möglichen Kontamination zu achten.
Die Hämofiltrationslösung darf nur dann angewendet werden, wenn Behältnis (Umverpackung und Zweikammerbeutel), Peelnaht und Konnektoren unbeschädigt und intakt sind und wenn die Lösung klar und farblos und frei von sichtbaren Partikeln ist. Die Lösung darf nur nach Öffnen der Peelnaht und Mischen der beiden Lösungen angewendet werden. Weitere Hinweise siehe Abschnitt 6.6.
In Zweifelsfällen sollte der für die Behandlung zuständige Arzt über die Verwendung der Lösung entscheiden.
Die Hämofiltrationslösung sollte durch einen integrierten oder externen Wärmer auf etwa Körpertemperatur erwärmt werden. Unter keinen Umständen darf die Lösung infundiert werden, solange sie nicht Raumtemperatur erreicht hat.
Während der Anwendung dieses Arzneimittels wurden in seltenen Fällen Ausfällungen von weißem Calciumcarbonat in den Schläuchen beobachtet, insbesondere in Nähe der Pumpen- und der Heizungseinheit. Die Lösung in den Schläuchen sollte daher während der Hämofiltration alle 30 Minuten sorgfältig visuell überprüft werden, um sicherzustellen, dass die Lösung im Schlauchsystem klar und frei von Ausfällungen ist. Ausfällungen können auch mit erheblicher Verzögerung nach Behandlungsbeginn auftreten. Bei Auftreten von Ausfällungen müssen die Lösung und die Schläuche sofort ausgetauscht und der Patient sorgfältig überwacht werden.
4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Während der Behandlung kann es zu einer Abnahme der Konzentration von filtrierbaren Arzneimitteln im Blut kommen, z. B. bei Arzneimitteln mit geringer Proteinbindungskapazität. Falls erforderlich, sollte eine entsprechende korrigierende Therapie erfolgen.
Durch korrekte Dosierung der Hämofiltrationslösung und genaue Überwachung der klinisch-chemischen Parameter und der Vitalzeichen können Wechselwirkungen mit anderen Arzneimitteln vermieden werden.
Folgende Wechselwirkungen sind jedoch denkbar:
• Elektrolytsubstitutionen, parenterale Ernährung und andere Infusionen, die im Rahmen einer
intensivmedizinischen Behandlung üblicherweise angewendet werden, sowie Serumzusammensetzung und Flüssigkeitsstatus des Patienten beeinflussen sich gegenseitig. Dies muss bei der Verordnung einer Hämofiltrationsbehandlung berücksichtigt werden.
• Toxische Wirkungen von Digitalis können durch Hyperkaliämie, Hypermagnesiämie und Hypokalzämie maskiert werden. Die Korrektur dieser Elektrolyte durch Hämofiltration kann Anzeichen und Symptome einer Digitalisvergiftung, z. B. Herzrhythmusstörungen, hervorrufen. Bei niedrigen Kaliumspiegeln oder hohen Calciumspiegeln kann es im Rahmen einer Digitalistherapie bei suboptimalen Dosen zu einer Digitalisvergiftung kommen.
• Vitamin D und calciumhaltige Arzneimittel, z. B. Calciumcarbonat als Phosphatbinder, können das Risiko einer Hyperkalzämie erhöhen.
• Zusätzliche Natriumbicarbonat-Substitution kann das Risiko einer metabolischen Alkalose erhöhen.
4.6 Fertilität, Schwangerschaft und Stillzeit
Schwangerschaft
Bisher liegen keine Erfahrungen mit der Anwendung von Duosol bei Schwangeren oder aus tierexperimentellen Studien vor. Da alle Bestandteile der Hämofiltrationslösung physiologische Substanzen sind, die der Substitution der bei der Hämofiltration entfernten essentiellen Plasmabestandteile dienen, sind jedoch keine Risiken für das ungeborene Kind zu erwarten. Falls notwendig, kann eine Anwendung von Duosol während der Schwangerschaft in Betracht gezogen werden.
Stillzeit
Da alle Bestandteile der Hämofiltrationslösung physiologische Substanzen sind, die der Substitution der bei der Hämofiltration entfernten essentiellen Plasmabestandteile dienen, sind keine Risiken für das Kind zu erwarten. Falls notwendig, kann eine Anwendung von Duosol während der Stillzeit in Betracht gezogen werden.
Fertilität
Da alle Bestandteile der Hämofiltrationslösung physiologische Substanzen sind, die der Substitution der bei der Hämofiltration entfernten essentiellen Plasmabestandteile dienen, sind keine Auswirkungen auf die Fertilität zu erwarten.
4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Nicht zutreffend.
4.8 Nebenwirkungen
Es sind keine Nebenwirkungen berichtet worden, die möglicherweise mit der bicarbonatgepufferten Hämofiltrationslösung in Zusammenhang gebracht werden können. Die folgenden Nebenwirkungen könnten jedoch Folge der Behandlung sein oder durch die verwendete Lösung hervorgerufen werden. Die Häufigkeit dieser Nebenwirkungen ist nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar):
Stoffwechsel- und Ernährungsstörungen
Hyperhydrierung oder Dehydratation, Elektrolytstörungen (z. B. Hyperkaliämie), Hypophosphatämie, Hyperglykämie, metabolische Alkalose
Gefäßerkrankungen Hypertonie, Hypotonie
Erkrankungen des Gastrointestinaltrakts Übelkeit, Erbrechen
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen Muskelkrämpfe
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels.
Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung über das in Anhang V aufgeführte nationale Meldesystem anzuzeigen.
4.9 Überdosierung
Bei bestimmungsgemäßer Dosierung ist das Auftreten von Notfallsituationen bisher nicht berichtet worden, zudem kann die Zufuhr der Lösung jederzeit unterbrochen werden. Bei ungenauer Bilanzierung kann es entweder zu einer Hyperhydrierung oder zu einer Dehydratation kommen. Diese manifestieren sich in Veränderungen des Blutdrucks, des zentralen Venendrucks, der Herzfrequenz und des Pulmonalarteriendrucks.
Bei einer Hyperhydrierung muss die Ultrafiltration erhöht und die Zufuhrgeschwindigkeit und das Volumen der Hämofiltrationslösung reduziert werden.
In Fällen einer starken Dehydratation muss die Ultrafiltration verringert oder beendet und das infundierte Volumen der Hämofiltrationslösung entsprechend erhöht werden.
Eine Bicarbonat-Überdosierung kann auftreten, wenn ein zu großes Volumen an Hämofiltrationslösung verabreicht wird. Dies kann zu metabolischer Alkalose, einer Verringerung von ionisiertem Calcium oder Tetanie führen.
Eine Überdosierung kann zu kongestivem Herzversagen und/oder Lungenstauung und zu Störungen der Elektrolytkonzentrationen und des Säure-Basen-Haushalts führen.
5. PHARMAKOLOGISCHE EIGENSCHAFTEN
5.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Hämofiltrate, ATC-Code: B05ZB Grundprinzipien der Hämofiltration
Während einer kontinuierlichen Hämofiltration werden Wasser und gelöste Substanzen wie urämische Toxine, Elektrolyte und Bicarbonat durch Ultrafiltration aus dem Blut entfernt. Das Ultrafiltrat wird durch eine Hämofiltrationslösung mit bilanzierten Elektrolyt- und Pufferkonzentrationen ersetzt.
Die gebrauchsfertige Lösung, bestehend aus einer Bicarbonat- und einer Elektrolytlösung, ist eine gemischte, bicarbonatgepufferte Hämofiltrationslösung zur Behandlung des akuten Nierenversagens mittels kontinuierlicher Hämofiltration.
Die Elektrolyte Na+, K+, Mg2+, Ca2+, Cl- und Bicarbonat sind für die Aufrechterhaltung und Korrektur der Flüssigkeits- und Elektrolythomöostase (Blutvolumen, osmotisches Gleichgewicht, Säure-Basen-Haushalt) unentbehrlich.
Die Wirksamkeit vergleichbarer intravenös angewendeter Lösungen zur Aufrechterhaltung des Säure-BasenGleichgewichts während der Hämofiltration hat sich in Studien und in langjährigem klinischem Einsatz zweifelsfrei erwiesen. Ihre Sicherheit und gute Verträglichkeit sind belegt. Die Pharmakologie von intravenös angewendeten Elektrolyten und Bicarbonat ist hinreichend bekannt.
5.2 Pharmakokinetische Eigenschaften
Die gebrauchsfertige Hämofiltrationslösung ist intravenös anzuwenden. Die Verteilung von Elektrolyten und Bicarbonat wird abhängig von Bedarf, metabolischen Bedingungen und residualer Nierenfunktion geregelt. Die Bestandteile der Hämofiltrationslösung werden mit Ausnahme von Glucose nicht metabolisiert. Die Ausscheidung von Wasser und Elektrolyten ist abhängig vom zellulären Bedarf, metabolischen Zustand, der residualen Nierenfunktion und von Flüssigkeitsverlusten z. B. über Darm, Lunge und Haut.
Da alle Bestandteile der Hämofiltrationslösung physiologische Substanzen sind, die der Substitution der bei der Hämofiltration entfernten essentiellen Plasmabestandteile dienen, wurden keine toxikologischen Studien durchgeführt.
6. PHARMAZEUTISCHE ANGABEN
6.1 Liste der sonstigen Bestandteile
Elektrolytlösung (kleine Kammer)
Salzsäure 25 % (zur pH-Einstellung)
Wasser für Injektionszwecke
Bicarbonatlösung (große Kammer)
Kohlendioxid (zur pH-Einstellung)
Wasser für Injektionszwecke
6.2 Inkompatibilitäten
Da keine Kompatibilitätsstudien durchgeführt wurden, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden. Wenn die Zugabe eines Arzneimittels zur Hämofiltrationslösung erforderlich ist, sollte es nur nach sorgfältiger Beurteilung seiner Kompatibilität mit der Hämofiltrationslösung und nur nach gründlicher Mischung der beiden Lösungen im Zweikammerbeutel erfolgen.
6.3 Dauer der Haltbarkeit
Dauer der Haltbarkeit im unversehrten Behältnis 2 Jahre
Dauer der Haltbarkeit nach Herstellung der gebrauchsfertigen Lösung
Die gemischte Lösung sollte sofort angewendet werden. Die gebrauchsfertige Zubereitung ist 24 Stunden bei 25 °C physikalisch und chemisch stabil.
6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Nicht über 30 °C lagern.
Nicht im Kühlschrank lagern oder einfrieren.
6.5 Art und Inhalt des Behältnisses
Zweikammerbeutel auf Polypropylen(PP)-Basis, der 4445 ml Bicarbonatlösung und 555 ml Elektrolytlösung enthält, welche durch eine Peelnaht voneinander getrennt sind, in einer Umverpackung auf PP-Basis. In der großen Kammer befinden sich zwei Schläuche auf PP-Basis, die durch Luer-Lock-Konnektoren auf Polycarbonat-Basis verschlossen sind. Der Schlauch in der kleinen Kammer wird nur bei der Herstellung verwendet und ist nicht zum Gebrauch gedacht.
2 Beutel zu 5000 ml (Zweikammerbeutel, 4445 ml und 555 ml) pro Umkarton
6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung
Hinweise zur Herstellung der gebrauchsfertigen Hämofiltrationslösung
Das Behältnis und die Lösung müssen vor Anwendung visuell überprüft werden. Die Hämofiltrationslösung darf nur dann angewendet werden, wenn Behältnis (Umverpackung und Zweikammerbeutel), Peelnaht und Konnektoren unbeschädigt und intakt sind und wenn die Lösung klar und farblos und frei von sichtbaren Partikeln ist.
Umverpackung erst unmittelbar vor der Anwendung entfernen
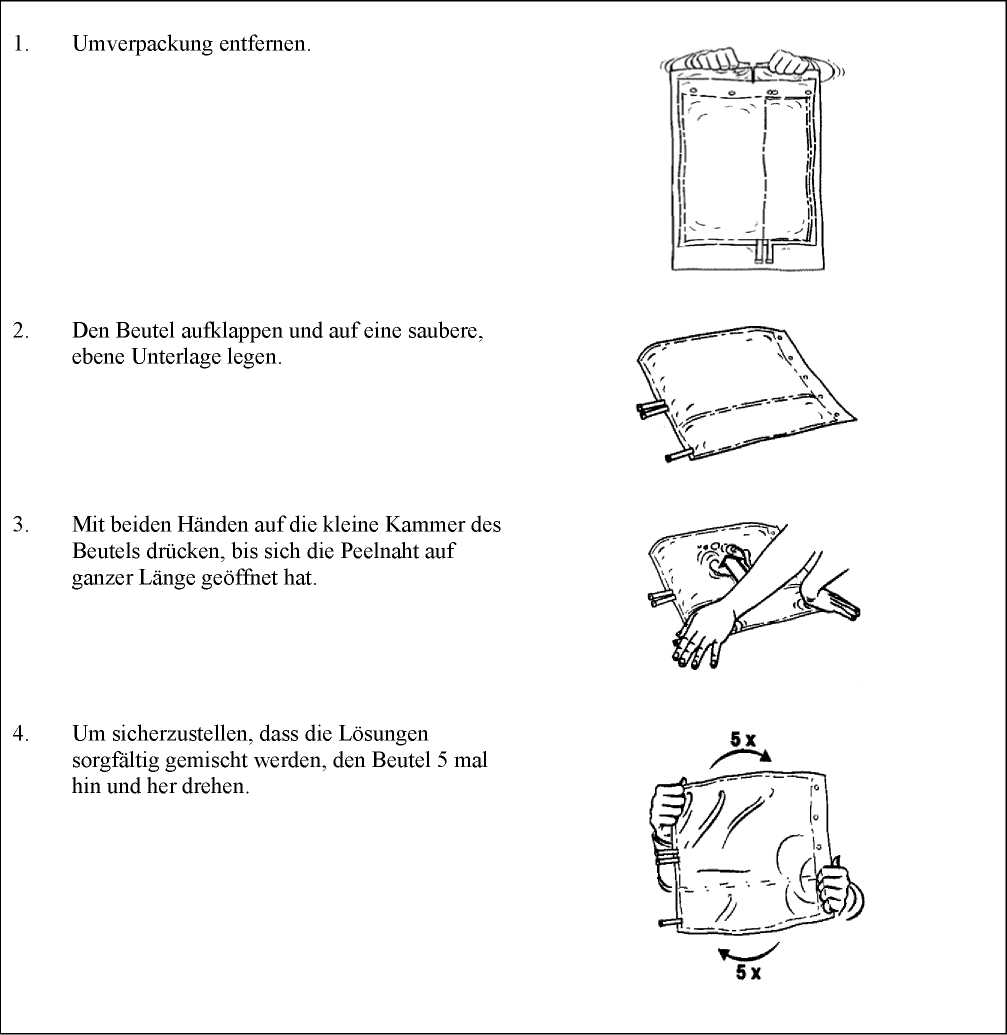
Nur zum einmaligen Gebrauch. Nicht verbrauchte Lösung und beschädigte Behältnisse sind zu verwerfen.
7. INHABER DER ZULASSUNG
B. Braun Avitum AG Schwarzenberger Weg 73-79 34212 Melsungen Deutschland
8. ZULASSUN GSNUMMER(N)
63372.00.00
9. DATUM DER ERTEILUNG DER ZULASSUNG/VERLÄNGERUNG DER ZULASSUNG
Datum der Erteilung der Zulassung: 09. Juni 2006
Datum der letzten Verlängerung der Zulassung: 19. Januar 2010
10. STAND DER INFORMATION
März 2015
11. VERKAUFSABGRENZUNG Apothekenpflichtig
8