Lemtrada
alt informationenANHANGI
ZUSAMMENFASSUNG DER MERKMALE DES ARZNEIMITTELS
▼ Dieses Arzneimittel miterliegt einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden. Hinweise zur Meldung von Nebenwirkungen, siehe Abschnitt 4.8.
1. BEZEICHNUNG DES ARZNEIMITTELS
LEMTRADA 12 mg Konzentrat zur Herstellung einer Infusionslösung.
2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
Jede Durchstechflasche enthält 12 mg Alemtuzumab in 1,2 ml (10 mg/ml).
Alemtuzumab ist ein monoklonaler Antikörper, der in Säugerzellen (Ovarialzellen des chinesischen Hamsters) in einem Nährmedium als Suspensionskultur mithilfe rekombinanter DNS-Technologie hergestellt wird.
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
3. DARREICHUNGSFORM
Konzentrat zur Herstellung einer Infusionslösung (steriles Konzentrat).
Ein klares, farbloses bis leicht gelbes Konzentrat mit einem pH-Wert von 7,0 bis 7,4.
4. KLINISCHE ANGABEN
4.1 Anwendungsgebiete
LEMTRADA ist angezeigt zur Behandlung von erwachsenen Patienten mit schubförmig-remittierender Multipler Sklerose (RRMS) mit aktiver Erkrankung, definiert durch klinischen Befund oder Bildgebung (siehe Abschnitte 4.4 und 5.1).
4.2 Dosierung und Art der Anwendung
Die Einleitung und die Überwachung der Behandlung mit LEMTRADA sollten durch einen in der Behandlung von MS-Patienten erfahrenen Neurologen erfolgen. Es sollten zudem Fachpersonal und Geräte zur Verfügung stehen, die geeignet sind, die häufigsten Nebenwirkungen, insbesondere Autoimmunerkrankungen und Infektionen, rechtzeitig zu erkennen und zu beherrschen.
Medikamente und Ausrüstung zur Behandlung von Überempfindlichkeitsreaktionen und/oder anaphylaktischen Reaktionen sollten verfügbar sein.
Mit LEMTRADA behandelten Patienten müssen die Patientenkarte und der Leitfaden für Patienten ausgehändigt werden. Außerdem müssen sie über die Risiken von LEMTRADA aufgeklärt werden (siehe auch die Packungsbeilage).
Dosierung
Die empfohlene Dosis von LEMTRADA beträgt 12 mg/Tag, verabreicht als intravenöse Infusion in 2 Behandlungsphasen:
• Behandlungsphase im ersten Behandlungsjahr:12 mg/Tag an 5 aufeinander folgenden Tagen (60 mg Gesamtdosis)
• Behandlungsphase im zweiten Behandlungsjahr: 12 mg/Tag an 3 aufeinander folgenden Tagen (36 mg Gesamtdosis), verabreicht 12 Monate nach der ersten Behandlungsphase.
Ausgelassene Dosen sollten nicht am gleichen Tag wie eine geplante Dosis verabreicht werden.
Nachbeobachtung von Patienten
Die Therapieempfehlung umfasst 2 Behandlungsphasen (siehe Abschnitt Dosierung) sowie die Nachbeobachtung der Patienten ab Behandlungsbeginn bis 48 Monate nach der letzten Infusion (siehe Abschnitt 4.4.).
Vorbehandlung
Die Patienten sollten an jedem der ersten 3 Tage einer jeden Behandlungsphase unmittelbar vor der Verabreichung von LEMTRADA mit Kortikosteroiden vorbehandelt werden. In klinischen Studien wurden die Patienten an den ersten 3 Tagen einer jeden Behandlungsphase mit LEMTRADA mit 1.000 mg Methylprednisolon vorbehandelt.
Zusätzlich kann auch eine Vorbehandlung mit Antihistaminika und/oder Antipyretika vor der Verabreichung von LEMTRADA in Erwägung gezogen werden.
Eine orale Prophylaxe gegen Herpes-Infektionen sollte bei allen Patienten durchgeführt werden. Die Prophylaxe sollte am ersten Tag einer jeden Behandlungsphase mit LEMTRADA beginnen und mindestens 1 Monat über den Abschluss der jeweiligen Behandlungsphase hinaus fortgeführt werden (siehe auch „Infektionen“ in Abschnitt 4.4). In klinischen Studien wurde den Patienten zweimal täglich 200 mg Aciclovir oder ein äquivalentes Arzneimittel verabreicht.
Ältere Personen
In den klinischen Studien waren keine Patienten im Alter über 55 Jahre eingeschlossen. Daher wurde nicht bestimmt, ob sie anders als jüngere Patienten auf die Behandlung ansprechen.
Patienten mit Nieren- oder Leberfunktionsbeeinträchtigung
LEMTRADA wurde bei Patienten mit Nieren- oder Leberfunktionsbeeinträchtigung nicht untersucht.
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von LEMTRADA bei Kindern und Jugendlichen mit MS im Alter von 0 bis 18 Jahren ist noch nicht belegt. Die Anwendung von Alemtuzumab bei Kindern im Alter von der Geburt bis unter zehn Jahren zur Behandlung von Multipler Sklerose ist nicht angezeigt. Es liegen keine Daten vor.
Art der Anwendung
LEMTRADA muss vor der Infusion verdünnt werden. Die verdünnte Lösung soll als intravenöse Infusion über einen Zeitraum von etwa 4 Stunden verabreicht werden.
Hinweise zur Verdünnung des Arzneimittels vor der Anwendung, siehe Abschnitt 6.6.
4.3 Gegenanzeigen
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile. Human-Immunodeficiency-Virus-Infektion (HIV-Infektion).
4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Die Anwendung von LEMTRADA wird nicht empfohlen bei Patienten, die keine aktive Erkrankung aufweisen oder unter der aktuellen Therapie stabil sind.
Mit LEMTRADA behandelten Patienten müssen die Packungsbeilage, die Patientenkarte und der Leitfaden für Patienten ausgehändigt werden. Vor der Behandlung müssen die Patienten über die Risiken und den Nutzen der Behandlung, sowie die Notwendigkeit einer 48-monatigen Nachbeobachtung nach der letzten LEMTRADA-Infusion aufgeklärt werden.
Autoimmunität
Die Behandlung kann zur Bildung von Autoantikörpern und einem erhöhten Risiko für autoimmun vermittelte Erkrankungen, einschließlich idiopathische thrombozytopenische Purpura (ITP), Schilddrüsenerkrankungen oder in seltenen Fällen Nephropathien (z. B. Goodpasture-Syndrom), führen. Bei Patienten mit anderen anamnestischen Autoimmunerkrankungen als MS ist besondere Vorsicht geboten, auch wenn verfügbare Daten nahe legen, dass nach einer Behandlung mit Alemtuzumab keine Verschlechterung von existierenden Autoimmunerkrankungen zu erwarten ist.
Idiopathische thrombozytopenische Purpura (ITP)
Schwerwiegende Ereignisse von ITP wurden bei etwa 1% der behandelten Patienten in kontrollierten klinischen Studien zu MS beobachtet. In einer kontrollierten klinischen Studie bei Patienten mit MS entwickelte ein Patient vor der Einführung der Auflage monatlicher Blutuntersuchungen eine ITP, die unerkannt blieb. Der Patient starb an einer Hirnblutung. ITP trat im Allgemeinen 14 bis 36 Monate nach der ersten Exposition auf. Symptome einer ITP umfassen unter anderem (aber nicht ausschließlich) eine erhöhte Neigung zu Blutergüssen, Petechien, spontane Schleimhautblutungen (z. B. Epistaxis, Hämoptyse), stärkere oder unregelmäßige Menstruationsblutungen. Hämoptyse kann auch ein Symptom des Goodpasture-Syndroms sein (siehe unten), weswegen eine entsprechende Differentialdiagnose erforderlich ist. Erinnern Sie den Patienten daran, auf mögliche Symptome zu achten und ärztliche Hilfe aufzusuchen, wenn Fragen bestehen.
Vor Beginn der Behandlung und danach monatlich bis 48 Monate nach der letzten Infusion sollte ein großes Blutbild mit Differentialblutbild erstellt werden. Danach sollten Untersuchungen durchgeführt werden, wenn die klinischen Befunde auf eine ITP hindeuten. Wenn ein Verdacht auf eine ITP besteht, sollte unverzüglich ein großes Blutbild erstellt werden.
Wenn das Vorliegen einer ITP bestätigt wird, sollten umgehend entsprechende medizinische Maßnahmen eingeleitet werden, einschließlich der sofortigen Überweisung an einen Facharzt. Daten aus klinischen MS-Studien haben gezeigt, dass das Erfüllen der Auflage monatlicher Blutuntersuchungen und Schulungen zu Zeichen und Symptomeneiner ITP zu einer frühzeitigen Erkennung und Behandlung von ITP verhelfen, sodass in den meisten Fällen auf die Erstlinienbehandlung angesprochen wird.
Das potentielle Risiko, das mit einer Wiederaufnahme der Behandlung mit LEMTRADA nach Auftreten einer ITP assoziiert ist, ist unbekannt.
Nephropathien
Nephropathien, einschließlich Goodpasture-Syndrom (anti-GBM-Glomerulonephritis), wurden bei 0,3% der Patienten in klinischen Studien zu MS beobachtet und traten im Allgemeinen innerhalb von 39 Monaten nach der letzten Verabreichung von LEMTRADA auf. In klinischen Studien traten 2 Fälle von Goodpasture-Syndrom auf. Beide Fälle waren schwerwiegend, wurden durch die klinische und Laborüberwachung früh erkannt und hatten nach Behandlung einen positiven Ausgang.
Die klinischen Merkmale einer Nephropathie können eine Erhöhung des Kreatinins im Blut, Hämaturie und/oder Proteinurie umfassen. Obwohl dies in klinischen Studien nicht beobachtet wurde, kann eine alveoläre Blutung, die sich als Hämoptyse äußert, im Rahmen des Goodpasture-Syndroms auftreten. Da Hämoptysen auch Symptom einer ITP sein können(siehe oben), ist eine entsprechende Differentialdiagnostik erforderlich. Der Patient sollte daran erinnert werden, auf mögliche Symptome zu achten und ärztliche Hilfe aufzusuchen, wenn Fragen bestehen. Das Goodpasture-Syndrom kann zu Nierenversagen führen, welches bei zu spät einsetzender Behandlung zu Dialysepflicht führt und/oder eine Transplantation erfordert und nicht behandelt lebensbedrohlich verlaufen kann.
Vor Beginn der Behandlung und danach monatlich bis 48 Monate nach der letzten Infusion sollten die Serum-Kreatinin-Spiegel überwacht werden. Urinuntersuchungen einschließlich Mikroskopie sollten vor Beginn der Behandlung und danach monatlich bis 48 Monate nach der letzten Infusion durchgeführt werden. Bei Beobachtung klinisch signifikanter Veränderungen ausgehend von den Ausgangswerten beim SerumKreatinin, bei ungeklärter Hämaturie und/oder Proteinurie sollten unverzüglich weitere Untersuchungen im Hinblick auf mögliche Nephropathien veranlasst werden, einschließlich einer sofortigen Überweisung an einen Facharzt. Die frühzeitige Erkennung und Behandlung von Nephropathien können das Risiko nicht erfolgreicher Behandlungsergebnisse senken. Nach diesem Zeitraum sollten Untersuchungen auf der Grundlage von klinischen Befunden, die eine Nephropathie nahelegen, durchgeführt werden.
Das potentielle Risiko, das mit einer Wiederaufnahme der Behandlung mit LEMTRADA nach Auftreten von Nephropathien assoziiert ist, ist unbekannt.
Schilddrüsenerkrankungen
Autoimmune Schilddrüsenerkrankungen wurden bei schätzungsweisen 36% der im Rahmen klinischer MS-Studien mit LEMTRADA 12 mg behandelten Patienten in den 48 Monaten nach der ersten Exposition mit LEMTRADA beobachtet. Bei Patienten mit anamnestischen Schilddrüsenerkrankungen war die Inzidenz von Schilddrüsenereignissen, sowohl in der mit LEMTRADA behandelten Gruppe, als auch in der mit Interferon beta-1a (IFNB-1a) behandelten Gruppe, höher. Patienten mit bestehender Schilddrüsenerkrankung sollte LEMTRADA nur verabreicht werden, wenn der mögliche Nutzen die möglichen Risiken überwiegt. Zu den beobachteten autoimmunen Schilddrüsenerkrankungen zählten Hyper- bzw. Hypothyreosen. Die meisten Ereignisse waren von leichtem bis mittlerem Schweregrad. Vor der Zulassung traten schwerwiegende Ereignisse bei < 1% der Patienten auf, wobei ausschließlich die Basedow-Krankheit (die auch als Graves' Disease bekannt ist), sowie Hyper- und Hypothyreosen bei mehr als einem Patienten auftraten. Die meisten Schilddrüsen-Ereignisse wurden mit konventionellen Therapien behandelt. Einige Patienten benötigten jedoch einen operativen Eingriff. In klinischen Studien durften Patienten, bei denen Schilddrüsen-Ereignisse festgestellt wurden, erneut mit LEMTRADA behandelt werden. Bisher liegen zwar nur begrenzte Erfahrungen vor, jedoch kam es im Allgemeinen bei erneut behandelten Patienten nicht zu einer Erhöhung des Schweregrads der Schilddrüsenerkrankung. Die Fortsetzung der Behandlung mit LEMTRADA sollte deswegen in jedem einzelnen Fall unter Berücksichtigung des klinischen Zustands des Patienten erwogen werden.
Vor Beginn der Behandlung und danach alle 3 Monate bis 48 Monate nach der letzten Infusion sollten Schilddrüsenfunktionstests, z.B. eine Bestimmung des Thyreoidea-stimulierenden Hormons (TSH), durchgeführt werden. Nach dieser Zeit sollten entsprechende Tests auf der Grundlage klinischer Befunde, die eine Schilddrüsenfunktionsstörung nahelegen, durchgeführt werden.
Eine Schilddrüsenerkrankung ist ein spezielles Risiko bei schwangeren Frauen (siehe Abschnitt 4.6).
In klinischen Studien war der Anti-Thyreoperoxidase-(Anti-TPO)-Antikörper-Wert eines Patienten vor der Behandlung nicht indikativ für das Aufftreten Schilddrüsen-assoziierter Nebenwirkungen. Bei der Hälfte der Patienten, die zu Studienbeginn Anti-TPO-Antikörper-positiv waren, und bei einem Viertel der Patienten, die zu Studienbeginn Anti-TPO-Antikörper-negativ waren, trat ein Schilddrüsen-Ereignis auf. Die große Mehrheit (etwa 80%) der Patienten, die nach der Behandlung mit einem Schilddrüsen-Ereignis vorstellig wurden, waren zu Studienbeginn Anti-TPO-Antikörper-negativ. Demzufolge können SchilddrüsenNebenwirkungen ungeachtet des Anti-TPO-Antikörper-Werts vor der Behandlung, auftreten, weswegen alle vorstehend beschriebenen Tests regelmäßig durchzuführen sind.
Zytopenien
Verdachtsfälle autoimmuner Zytopenien, wie etwa Neutropenien, hämolytische Anämien und Panzytopenien, wurden in klinischen MS-Studien selten berichtet. Die Ergebnisse des großen Blutbildes (siehe oben unter ITP) sollten genutzt werden, um die Patienten hinsichtlich des Auftretens von Zytopenien zu überwachen. Wenn das Vorliegen einer Zytopenie bestätigt wird, sollten umgehend entsprechende medizinische Maßnahmen eingeleitet werden, einschließlich der Überweisung an einen Facharzt.
Infusionsassoziierte Reaktionen (IAR)
In kontrollierten klinischen Studien waren infusionsassoziierte Reaktionen (IAR) als jedes unerwünschte Ereignis definiert, das während oder innerhalb von 24 Stunden nach der LEMTRADA-Infusion auftrat. Die meisten dieser Ereignisse könnten auf eine Zytokinfreisetzung während der Infusion zurückzuführen sein. Die meisten der im Rahmen klinischer MS-Studien mit LEMTRADA behandelten Patienten entwickelten leichte bis mittelschwere IAR während und/oder bis zu 24 Stunden nach der Verabreichung von LEMTRADA 12 mg. Hierzu zählten oft Kopfschmerz, Ausschlag, Fieber, Übelkeit, Urtikaria, Pruritus, Schlaflosigkeit, Schüttelfrost, Hitzegefühl, Ermüdung, Dyspnoe, Geschmacksstörung, Beklemmungsgefühl in der Brust, generalisierter Ausschlag, Tachykardie, Bradykardie, Dyspepsie, Schwindelgefühl und Schmerz. Schwerwiegende Reaktionen traten bei 3% der Patienten auf. Hierzu zählten Fälle von Fieber, Urtikaria, Vorhofflimmern, Übelkeit, Beklemmungsgefühl in der Brust und Hypotonie. Die klinischen Merkmale von anaphylaktischen Reaktionen können den klinischen Merkmalen von infusionsassoziierten Reaktionen ähneln, sind aber in der Regel schwerwiegender und potentiell lebensbedrohlich. Im Gegensatz zu infusionsassoziierten Reaktionen wurde über anaphylaktische Reaktionen selten berichtet.
Es wird empfohlen, die Patienten vorzubehandeln, um infusionsassoziierte Reaktionen zu lindern (siehe Abschnitt 4.2). Die meisten Patienten in kontrollierten klinischen Studien erhielten Antihistaminika und/oder Antipyretika vor mindestens einer LEMTRADA-Infusion. IAR können bei Patienten trotz Vorbehandlung auftreten. Eine Überwachung hinsichtlich infusionsassoziierter Reaktionen wird während und für weitere 2 Stunden nach Beendigung der LEMTRADA-Infusion empfohlen. Falls eine IAR auftritt, muss nach Bedarf eine entsprechende symptomatische Behandlung eingeleitet werden. Wenn die Infusion nicht gut vertragen wird, kann die Infusionsdauer verlängert werden. Wenn eine schwerwiegende Infusionsreaktion auftritt, sollte der unverzügliche Abbruch der intravenösen Infusion erwogen werden. In den klinischen Studien waren anaphylaktische oder andere schwerwiegende Reaktionen, die einen Behandlungsabbruch erforderten, sehr selten. Ärzte sollten die kardiologische Anamnese des Patienten kennen, da auch kardiale Symptome wie Tachykardie zu den infusionsassoziierten Reaktionen gehören.
Medikamente und Ausrüstung zur Behandlung anaphylaktischer und/oder schwerwiegender Reaktionen sollten verfügbar sein.
Infektionen
Infektionen traten in kontrollierten klinischen Studien zu MS, die bis zu 2 Jahre lang andauerten, bei 71% der mit LEMTRADA 12 mg behandelten Patienten im Vergleich zu 53% der mit subkutan verabreichtem Interferon Beta-1a [IFNB-1a] (44 pg, 3-mal wöchentlich) behandelten Patienten auf und waren überwiegend von leichtem bis mittlerem Schweregrad. Infektionen, die häufiger bei mit LEMTRADA behandelten Patienten als bei mit IFNB-1a behandelten Patienten auftraten, waren Nasopharyngitis,
Harnwegsinfektionen, Infektionen der oberen Atemwege, Sinusitis, oraler Herpes, Grippe und Bronchitis. Schwerwiegende Infektionen traten in kontrollierten klinischen Studien zu MS bei 2,7% der mit LEMTRADA behandelten Patienten im Vergleich zu 1% der mit IFNB-1a behandelten Patienten auf. Schwerwiegende Infektionen in der LEMTRADA-Gruppe umfassten: Appendizitis, Gastroenteritis, Pneumonie, Herpes zoster und Zahninfektionen. Die Infektionen waren im Allgemeinen von typischer Dauer und bildeten sich nach konventioneller medizinischer Behandlung zurück.
Schwerwiegende Infektionen mit dem Varizella-Zoster-Virus, einschließlich primärer Varizella-Infektion (Windpocken) und Reaktivierung des Varizella-Zoster-Virus (Herpes zoster), traten in klinischen Studien häufiger bei mit LEMTRADA 12 mg behandelten Patienten (0,3%) als bei mit IFNB-1a behandelten Patienten (0%) auf. Zervikale Infektionen mit dem humanen Papillomavirus (HPV), einschließlich Zervixdysplasie, wurden bei mit LEMTRADA 12 mg behandelten Patienten ebenfalls berichtet (2%). Es wird empfohlen, bei weiblichen Patienten jährlich ein HPV-Screening durchzuführen.
Das Auftreten einer Tuberkulose wurde in kontrollierten klinischen Studien bei mit LEMTRADA und bei mit IFNB-1a behandelten Patienten berichtet. Eine aktive und latente Tuberkulose wurden bei 0,3% der mit LEMTRADA behandelten Patienten berichtet, meistens in endemischen Regionen. Vor Beginn der Behandlung müssen alle Patienten sowohl auf aktive als auch inaktive (latente) Tuberkulose gemäß den lokalen Richtlinien untersucht werden.
Bei Patienten, die mit LEMTRADA behandelt wurden, wurde das Auftreten einer
Listeriose/Listerienmeningitis berichtet, die meist innerhalb eines Monats nach der LEMTRADA-Infusion auftrat. Um dieses Risiko zu reduzieren, sollten Patienten, die LEMTRADA erhalten, die Aufnahme von rohem oder nicht durchgegartem Fleisch, Weichkäse und unpasteurisierten Milchprodukten bis mindestens einen Monat nach der LEMTRADA-Behandlung vermeiden.
Oberflächliche Pilzinfektionen, insbesondere orale und vaginale Candidosen, traten in kontrollierten klinischen Studien zu MS häufiger bei mit LEMTRADA behandelten Patienten (12%) als bei mit IFNB-1a behandelten Patienten (3%) auf.
Ärzte sollten in Erwägung ziehen, den Beginn der Verabreichung von LEMTRADA bei Patienten mit aktiver Infektion zu verschieben, bis die Infektion vollständig kontrolliert ist.
Eine Prophylaxe mit oralen Antiherpetika sollte bei allen Patienten durchgeführt werden. Die Prophylaxe sollte am ersten Tag der jeweiligen Behandlungsphase mit LEMTRADA beginnen und mindestens 1 Monat über den Abschluss der Behandlungsphase hinaus fortgeführt werden. In klinischen Studien wurde den Patienten zweimal täglich 200 mg Aciclovir oder ein äquivalentes Arzneimittel verabreicht.
LEMTRADA wurde bei der Behandlung von MS nicht gleichzeitig mit oder nach antineoplastischen oder immunsuppressiven Arzneimitteln verabreicht. Wie bei anderen immunmodulierenden Therapien sollte auch bei der Erwägung einer LEMTRADA-Gabe eine mögliche Kombinationswirkung auf das Immunsystem des Patienten in Betracht gezogen werden. Die gleichzeitige Anwendung von LEMTRADA und solchen Arzneimitteln könnte das Risiko einer Immunsuppression erhöhen.
Es liegen keine Daten zu einem möglichen Zusammenhang zwischen der Gabe von LEMTRADA und einer Reaktivierung des Hepatitis-B-Virus (HBV) oder des Hepatitis-C-Virus (HCV) vor, da Patienten mit Anzeichen aktiver oder chronischer Infektionen von den klinischen Studien ausgeschlossen wurden. Es sollte in Erwägung gezogen werden, Patienten mit hohem Risiko für eine HBV- und/oder HCV-Infektion vor Beginn der Behandlung mit LEMTRADA auf das Vorliegen einer solchen Infektion zu untersuchen. Bei der Verschreibung von LEMTRADA an Patienten, die als Träger von HBV und/oder HCV identifiziert wurden, ist Vorsicht geboten, da diese Patienten infolge ihres Status ein erhöhtes Risiko tragen, durch eine potentielle Virusreaktivierung irreversible Leberschäden davonzutragen.
Bösartige Neubildungen
Wie bei anderen immunmodulierenden Therapien ist auch bei Aufnahme einer LEMTRADA-Therapie bei Patienten mit anamnestischen oder noch bestehenden bösartigen Neubildungen Vorsicht geboten. Es ist derzeit nicht bekannt, ob Alemtuzumab das Risiko für die Entwicklung von bösartigen Schilddrüsenneubildungen erhöht, da eine Schilddrüsen-Autoimmunität selbst ein Risikofaktor für bösartige Schilddrüsenneubildungen sein kann.
Verhütung
Es wurden ein Plazentatransfer und eine potentielle pharmakologische Wirkung von LEMTRADA bei Mäusen während der Gestation und nach der Geburt beobachtet. Frauen im gebärfähigen Alter sollten während und 4 Monate lang nach einer Behandlungsphase mit LEMTRADA eine zuverlässige Verhütungsmethode anwenden (siehe Abschnitt 4.6).
Impfstoffe
Es wird empfohlen, dass Patienten die regionalen Impfanforderungen mindestens 6 Wochen vor Aufnahme der Behandlung mit LEMTRADA erfüllt haben. Die Fähigkeit, nach Behandlung mit LEMTRADA eine Immunantwort auf einen Impfstoff zu entwickeln, wurde nicht untersucht.
Die Sicherheit einer Immunisierung mit viralen Lebendimpfstoffen nach einer Behandlungsphase mit LEMTRADA wurde nicht formal in kontrollierten klinischen Studien zu MS untersucht. Virale Lebendimpfstoffe sollten nicht an MS-Patienten verabreicht werden, die kürzlich eine Behandlungsphase mit LEMTRADA erhalten haben.
Varizella-Zoster-Virus-Antikörper-Test/-Impfung
Wie bei allen immunmodulierenden Arzneimitteln sollten Patienten, die keine Windpocken in der Anamnese aufweisen oder nicht gegen das Varizella-Zoster-Virus (VZV) geimpft sind, vor Beginn einer Behandlungsphase mit LEMTRADA auf Antikörper gegen VZV getestet werden. Eine VZV-Impfung von antikörpernegativen Patienten sollte vor Beginn der Behandlung mit LEMTRADA in Erwägung gezogen werden. Um die vollständige Wirkung der VZV-Impfung zu ermöglichen, sollte die Behandlung mit LEMTRADA auf 6 Wochen nach der Impfung verschoben werden.
Empfohlene Laboruntersuchungen zur Patientenüberwachung
Die Laboruntersuchungen sollten in regelmäßigen Abständen über 48 Monate nach der letzten Behandlungsphase mit LEMTRADA durchgeführt werden, um die Patienten hinsichtlich früher Anzeichen einer Autoimmunerkrankung zu überwachen:
• Großes Blutbild mit Differentialblutbild (vor Beginn der Behandlung und danach in monatlichen Abständen)
• Kreatinin-Spiegel im Serum (vor Beginn der Behandlung und danach in monatlichen Abständen)
• Urinanalyse mit Mikroskopie (vor Beginn der Behandlung und danach in monatlichen Abständen)
• ein Schilddrüsenfunktionstest, wie etwa eine Bestimmung des Thyreotropin-Spiegels (vor Beginn der Behandlung und danach alle 3 Monate)
Nach diesem Zeitraum sind bei jedem klinischen Befund, der Nephropathien oder Schilddrüsenerkrankungen nahelegt, weitere Untersuchungen erforderlich.
Informationen aus der Anwendung von Alemtuzumab vor der Genehmigung für das Inverkehrbringen von LEMTRADA außerhalb von Studien, die vom Unternehmen finanziert wurden.
Die folgenden Nebenwirkungen wurden vor der Zulassung von LEMTRADA während der Anwendung von Alemtuzumab zur Behandlung der chronischen lymphatischen Leukämie vom B-Zell-Typ (B-CLL), sowie zur Behandlung anderer Erkrankungen, im Allgemeinen bei höheren und häufigeren Dosen (z. B. 30 mg), als in der empfohlenen Dosis zur Behandlung von MS, festgestellt. Da diese Reaktionen freiwillig von einer Population unbestimmter Größe berichtet wurden, ist es nicht immer möglich, ihre Häufigkeit zuverlässig zu schätzen oder einen Kausalzusammenhang zur Exposition mit Alemtuzumab herzustellen.
Autoimmunerkrankung
Zu den Autoimmunereignissen, die bei mit Alemtuzumab behandelten Patienten berichtet wurden, zählen Neutropenie, hämolytische Anämie (einschließlich eines Falls mit tödlichem Ausgang), erworbene Hämophilie, Goodpasture-Syndrom und Schilddrüsenerkrankung. Schwerwiegende und manchmal tödliche Autoimmunphänomene, einschließlich autoimmunhämolytischer Anämie, Autoimmunthrombozytopenie, aplastischer Anämie, Guillain-Barre-Syndrom und chronischer entzündlicher demyelinisierender Polyradikuloneuropathie, sind bei mit Alemtuzumab behandelten Patienten, die nicht an MS litten, berichtet worden. Ein positiver Coombs-Test ist bei mit Alemtuzumab behandelten Onkologie-Patienten beobachtet worden. Eine tödliche transfusionsassoziierte Graft-versus-Host-Erkrankung wurde bei einem mit Alemtuzumab behandelten Onkologie-Patienten berichtet.
Infusionsassoziierte Reaktionen
Schwerwiegende und manchmal tödliche IAR, einschließlich Bronchospasmus, Hypoxie, Synkope, Lungeninfiltrate, akutes Atemnotsyndrom, Atemstillstand, Myokardinfarkt, Arrhythmien, akute Herzinsuffizienz und Herzstillstand, wurden bei Patienten beobachtet, die nicht an MS litten und mit höheren und häufigeren Dosen von Alemtuzumab (als bei MS angewendet) behandelt wurden. Schwere anaphylaktische Reaktionen und andere Überempfindlichkeitsreaktionen, einschließlich anaphylaktischen Schocks und Angioödem, wurden ebenfalls berichtet.
Infektionen und parasitäre Erkrankungen
Schwerwiegende und manchmal tödliche virale, bakterielle, Protozoen- und Pilzinfektionen, einschließlich solcher infolge einer Reaktivierung latenter Infektionen, wurden bei Patienten berichtet, die nicht an MS litten und mit höheren und häufigeren Dosen von Alemtuzumab (als bei MS angewendet) behandelt wurden. Progressive multifokale Leukoenzephalopathien (PML) wurden bei Patienten mit B-CLL mit oder ohne Behandlung mit Alemtuzumab berichtet. Die Häufigkeit von PML bei mit Alemtuzumab behandelten B-CLL-Patienten ist nicht höher als die Hintergrundhäufigkeit.
Erkrankungen des Blutes und des Lymphsystems
Es wurde über schwere Blutungsreaktionen bei Patienten, die nicht an MS litten, berichtet.
Herzerkrankungen
Das Auftreten einer kongestiven Herzinsuffizienz, einer Kardiomyopathie und einer verkleinerten Auswurffraktion wurde bei mit Alemtuzumab behandelten Patienten, die nicht an MS litten und zuvor mit potentiell kardiotoxischen Substanzen behandelt wurden, berichtet.
Epstein-Barr-Virus assoziiertes lymphoproliferatives Syndrom
Fälle von Epstein-Barr-Virus assoziiertem lymphoproliferativem Syndrom wurden außerhalb von Studien, die vom Unternehmen finanziert wurden, beobachtet.
4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Es wurden keine formalen Wechselwirkungsstudien mit LEMTRADA unter Anwendung der empfohlenen Dosis für Patienten mit MS durchgeführt. In einer kontrollierten klinischen Studie zu MS mussten Patienten, die kürzlich mit beta-Interferon und Glatirameracetat behandelt worden waren, die Behandlungen 28 Tage vor Beginn der Behandlung mit LEMTRADA absetzen.
4.6 Fertilität, Schwangerschaft und Stillzeit
Frauen im gebärfähigen Alter
Die Serumspiegel innerhalb von 30 Tagen nach jeder Behandlungsphase waren niedrig oder nicht nachweisbare. Daher sollten Frauen im gebärfähigen Alter während und 4 Monate lang nach der Behandlungsphase eine zuverlässige Verhütungsmethode anwenden.
Schwangerschaft
Bisher liegen begrenzte Erfahrungen mit der Anwendung von LEMTRADA bei Schwangeren vor. LEMTRADA sollte während der Schwangerschaft nur verabreicht werden, wenn der potentielle Nutzen die potentiellen Risiken für den Fötus überwiegt.
Menschliches IgG passiert bekanntermaßen die Plazentaschranke; Alemtuzumab kann ebenfalls die Plazentaschranke überschreiten und dadurch ein potentielles Risiko für den Fötus darstellen. In Toxizitätsstudien an Tieren wurde Reproduktionstoxizität nachgewiesen (siehe Abschnitt 5.3). Es ist nicht bekannt, ob die Verabreichung von Alemtuzumab bei schwangeren Frauen zur Fruchtschädigung oder zur Einschränkung der Fruchtbarkeit führen kann.
Eine Schilddrüsenerkrankung (siehe Abschnitt 4.4 Schilddrüsenerkrankungen) stellt ein spezielles Risiko für schwangere Frauen dar. Ohne eine Behandlung der Hypothyreose während der Schwangerschaft besteht ein erhöhtes Risiko eines Spontanaborts und fötaler Auswirkungen, wie etwa geistige Retardierung und
Zwergwuchs. Bei Müttern mit Basedow-Krankheit können mütterliche Thyreotropin-Rezeptor-Antikörper auf einen in der Entwicklung befindlichen Fötus übertragen werden und eine vorübergehende neonatale Basedow-Krankheit zu Folge haben.
Stillzeit
Alemtuzumab wurde in der Milch und bei den Jungen säugender Mäuse nachgewiesen.
Es ist nicht bekannt, ob Alemtuzumab in die menschliche Milch übergeht. Ein Risiko für den gestillten Säugling kann nicht ausgeschlossen werden. Daher sollte das Stillen während einer Behandlungsphase von LEMTRADA und 4 Monate lang nach der letzten Infusion einer jeden Behandlungsphase unterbrochen werden. Allerdings kann der Nutzen der durch die Muttermilch übertragenen Immunität die Risiken einer potentiellen Exposition gegenüber Alemtuzumab für den gestillten Säugling überwiegen.
Fertilität
Bisher liegen keine hinreichenden klinischen Sicherheitsdaten zu den Auswirkungen von LEMTRADA auf die Fertilität vor. Eine Teilstudie mit 13 männlichen Patienten, die mit Alemtuzumab (entweder 12 mg oder 24 mg) behandelt wurden, gab keine Hinweise auf Aspermie, Azoospermie, beständig niedrige Spermienzahlen, Motilitätsstörungen oder einen Anstieg morphologischer Anomalien bei den Spermien.
CD52 ist bekanntermaßen im Reproduktionsgewebe des Menschen und von Nagetieren vorhanden. Daten aus Tierstudien haben Wirkungen auf die Fertilität bei humanisierten Mäusen (siehe Abschnitt 5.3) gezeigt; eine potentielle Wirkung auf die menschliche Fertilität während des Zeitraums der Exposition, basierend auf den verfügbaren Daten, ist jedoch nicht bekannt.
4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Es wurden keine Studien zu den Auswirkungen von LEMTRADA auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen durchgeführt.
Bei den meisten Patienten treten während oder innerhalb von 24 Stunden nach der Behandlung mit LEMTRADA infusionsassoziierte Reaktionen (IAR) auf. Einige IAR (z. B. Schwindelgefühl) können die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen vorübergehend beeinträchtigen, weswegen Vorsicht geboten ist, bis diese abgeklungen sind.
4.8 Nebenwirkungen
Zusammenfassung des Sicherheitsprofils
Insgesamt 1.188 Patienten mit schubförmig-remittierender Multipler Sklerose (RRMS), die mit LEMTRADA (entweder 12 mg oder 24 mg) behandelt wurden, bildeten die Sicherheitspopulation in einer gepoolten Analyse kontrollierter klinischer Studien, die 2.363 Patientenjahre Sicherheitsnachbeobachtung und einen medianen Nachbeobachtungszeitraum von 24 Monaten ergaben.
Die wichtigsten Nebenwirkungen sind Autoimmunität (ITP, Schilddrüsenerkrankungen, Nephropathien, Zytopenien), IAR und Infektionen. Diese sind unter Abschnitt 4.4 beschrieben.
Die häufigsten Nebenwirkungen unter LEMTRADA (bei > 20% der Patienten) sind Ausschlag, Kopfschmerz, Fieber und Atemwegsinfektionen.
Tabellarische Liste der Nebenwirkungen
Die folgende Tabelle basiert auf den gepoolten Sicherheitsdaten aus bis zu 24 Monaten von RRMS-Patienten, die zu Studienbeginn an 5 aufeinander folgenden Tagen und in Monat 12 der Studie an 3 aufeinander folgenden Tagen mit LEMTRADA 12 mg/Tag behandelt wurden. Nebenwirkungen, die bei > 0,5% der Patienten auftraten, sind gemäß der System-Organklassen (System Organ Class, SOC) und Preferred Term (PT; deutsch: Bevorzugte Bezeichnung) des Medical Dictionary for Regulatory Activities (MedDRA; deutsch: Medizinisches Wörterbuch für Aktivitäten im Rahmen der Arzneimittelzulassung) aufgeführt. Die Häufigkeiten sind wie folgt definiert: sehr häufig (> 1/10), häufig (> 1/100 bis < 1/10), gelegentlich (> 1/1.000 bis < 1/100). Innerhalb jeder Häufigkeitsgruppe wurden die Nebenwirkungen nach abnehmendem Schweregrad angegeben.
Tabelle 1: Nebenwirkungen in Studie 1, 2 und 3, die bei > 0,5% der mit LEMTRADA 12 mg behandelten Patienten beobachtet wurden
|
System-Organklasse |
Sehr häufig |
Häufig |
Gelegentlich |
|
Infektionen und parasitäre Erkrankungen |
Infektion der oberen Atemwege, Harnwegsinfektion |
Infektion der unteren Atemwege, Herpes zoster, Gastroenteritis, oraler Herpes, orale Candidose, vulvovaginale Candidose, Grippe, Ohreninfektion |
Zahninfektion, genitaler Herpes, Onychomykose |
|
Erkrankungen des Blutes und des Lymphsystems |
Lymphopenie, Leukopenie |
Lymphadenopathie |
Idiopatische thrombozytopenische Purpura (ITP), Thrombozytopenie, erniedrigte Hämoglobinwerte, erniedrigte Hämatokritwerte |
|
Erkrankungen des Immunsystems |
Zytokin-Freisetzungs Syndrom | ||
|
Endokrine Erkrankungen |
Basedow-Krankheit, Hyperthyreose, Immunthyreoiditis, Hypothyreose, Struma, positiver Schilddrüsenantikörpertest | ||
|
Psychiatrische Erkrankungen |
Schlaflosigkeit*, Ängstlichkeit |
Depression | |
|
Erkrankungen des Nervensystems |
Kopfschmerz* |
MS-Schub, Schwindelgefühl*, Hypoästhesie, Parästhesie, Tremor, Geschmacksstörung* |
Gefühlsstörung, Hyperästhesie |
|
Augenerkrankungen |
verschwommenes Sehen |
Konjunktivitis | |
|
Erkrankungen des Ohrs und des Labyrinths |
Vertigo | ||
|
Herzerkrankungen |
Tachykardie*, Bradykardie*, Palpitationen | ||
|
Gefäßerkrankungen |
Hitze gefühl* |
Hypotonie*, Hypertonie | |
|
Erkrankungen der Atemwege, des Brustraums und des Mediastinums |
Dyspnoe*, Husten, Epistaxis, Schmerzen im Oropharynx |
Engegefühl im Hals, Schluckauf, Rachenreizung | |
|
Erkrankungen des Gastrointestinaltrakts |
Übelkeit* |
Abdominalschmerz, Erbrechen, Diarrhoe, Dyspepsie*, Stomatitis |
Obstipation, gastroösophageale Refluxerkrankung, Zahnfleischbluten, |
|
Dysphagie | |||
|
Leber- und Gallenerkrankungen |
erhöhte Aspartataminotransferas e-Werte | ||
|
Erkrankungen der Haut und des Unterhautzellgewebes |
Urtikaria*, Ausschlag*, Pruritus* |
generalisierter Ausschlag *, Erythem, Ekchymose, Alopezie, Hyperhidrose, Akne |
Blasenbildung, nächtliche Schweißausbrüche |
|
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen |
Myalgie, Muskelschwäche, Arthralgie, Rückenschmerzen, Schmerz in einer Extremität, Muskelspasmen, Nackenschmerzen | ||
|
Erkrankungen der Nieren und Harnwege |
Proteinurie, Hämaturie | ||
|
Erkrankungen der Geschlechtsorgane und der Brustdrüse |
Menorrhagie, unregelmäßige Menstruation |
Zervixdysplasie, Amenorrhoe | |
|
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort |
Pyrexie*, Ermüdung* |
Beklemmungsgefühl in der Brust*, Schüttelfrost*, Schmerz*, periphere Ödeme, Asthenie, grippeähnliche Erkrankung, Unwohlsein, Schmerzen an der Infusionsstelle | |
|
Untersuchungen |
erniedrigtes Gewicht | ||
|
Verletzung, Vergiftung und durch Eingriffe bedingte Komplikationen |
Prellung |
Beschreibung ausgewählter Nebenwirkungen
Bei den mit einem Sternchen (*) versehenen Begriffen in Tabelle 1 handelt es sich um Nebenwirkungen, die als infusionsassoziierte Reaktionen (IAR) berichtet wurden. Zu den IAR zählen auch Vorhofflimmern und anaphylaktische Reaktionen, die unter der 0,5%-Grenze für arzneimittelbedingte Ereignisse liegen (siehe Abschnitt 4.4).
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung über das in Anhang V aufgeführte nationale Meldesystem anzuzeigen.
4.9 Überdosierung
In kontrollierten klinischen Studien erhielten zwei MS-Patienten unbeabsichtigt bis zu 60 mg LEMTRADA (d. h. die Gesamtdosis für die erste Behandlungsphase) in einer einzelnen Infusion. Sie erlitten schwerwiegende Reaktionen (Kopfschmerz, Ausschlag und entweder Hypotonie oder Sinustachykardie). Dosen von LEMTRADA, die über den in den klinischen Studien untersuchten Dosen liegen, können die Schwere und/oder Dauer infusionsassoziierter Reaktionen oder der Immunwirkungen erhöhen.
Es ist kein Antidot für Alemtuzumab-Überdosierungen bekannt. Die Behandlung besteht darin, die Arzneimittelgabe abzubrechen und für eine unterstützende Behandlung zu sorgen.
5. PHARMAKOLOGISCHE EIGENSCHAFTEN
5.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Selektive Immunsuppressiva, ATC-Code: L04AA34. Wirkmechanismus
Alemtuzumab ist ein rekombinanter, aus DNS abgeleiteter, humanisierter monoklonaler Antikörper, der sich gegen das 21- bis 28-kD-Glykoprotein CD52 auf der Zelloberfläche richtet. Alemtuzumab ist ein IgG1-Kappa-Antikörper mit humanem variablem Gerüst und konstanten Regionen und komplementärdeterminierenden Regionen eines murinen (Ratte) monoklonalen Antikörpers. Der Antikörper hat ein ungefähres Molekulargewicht von 150 kD.
Alemtuzumab bindet an CD52, ein Antigen auf der Zelloberfläche, das in hohen Konzentrationen auf T-Lymphozyten (CD3+) und B-Lymphozyten (CD19+) und in niedrigen Konzentrationen auf natürlichen Killerzellen, Monozyten und Makrophagen vorkommt. Es ist wenig oder kein CD52 auf Neutrophilen, Plasmazellen oder Knochenmark-Stammzellen nachweisbar. Alemtuzumab wirkt durch antikörperabhängige, zellvermittelte Zytolyse und komplementvermittelte Lyse nach Zelloberflächenbindung an T- und B-Lymphozyten.
Der Mechanismus, durch den LEMTRADA seine therapeutischen Wirkungen auf MS entfaltet, ist noch nicht vollständig aufgeklärt. Allerdings weist die Forschung in Richtung immunmodulatorischer Wirkungen durch die Depletion und Repopulation von Lymphozyten, einschließlich:
- Veränderungen in der Anzahl, den Anteilen und Eigenschaften einiger Lymphozytenuntergruppen nach der Behandlung
- Erhöhte Anteile an regulatorischen T-Zell-Untergruppen
- Erhöhte Anteile an Gedächtnis-T- und B-Lymphozyten
- Vorübergehende Wirkungen auf Bestandteile der angeborenen Immunität (d. h. Neutrophile, Makrophagen, NK-Zellen)
Die Senkung der Spiegel der zirkulierenden B- und T-Zellen durch LEMTRADA und die darauf folgende Repopulation können das Potential für einen Schub verkleinern, was letztlich die Progression der Erkrankung verzögert.
Pharmakodynamische Wirkungen
LEMTRADA führt nach jeder Behandlungsphase zu einer Depletion der zirkulierenden T- und B-Lymphozyten, wobei die niedrigsten beobachteten Werte einen Monat nach einer Behandlungsphase auftreten (der früheste Zeitpunkt nach der Behandlung in Phase-III-Studien). Es kommt im Laufe der Zeit zu einer Repopulation der Lymphozyten mit einer Erholung der B-Zellen, die in der Regel innerhalb von 6 Monaten abgeschlossen ist. Die CD3+- und CD4+-Lymphozytenzahlen steigen langsamer auf normale Werte. Im Allgemeinen erreichen sie die Ausgangswerte nicht bis 12 Monate nach der Behandlung. Bei etwa 40% der Patienten erreichten die Lymphozytengesamtzahlen 6 Monate nach jeder Behandlungsphase die untere Normgrenze (LLN) und bei etwa 80% der Patienten erreichten die Lymphozytengesamtzahlen 12 Monate nach jeder Behandlungsphase die LLN.
Die Anzahl der Neutrophilen, Monozyten, Eosinophilen, Basophilen und der natürlichen Killerzellen ist nur vorübergehend durch LEMTRADA verändert.
Klinische Wirksamkeit und Sicherheit
Die Sicherheit und Wirksamkeit von LEMTRADA wurde in 3 randomisierten, auswerterverblindeten klinischen Studien mit wirksamer Vergleichssubstanz bei Patienten mit RRMS untersucht.
Für die Studien 1 und 2 sind das Studiendesign/die Demographie der Studie und die Ergebnisse in Tabelle 2 und Tabelle 3 aufgeführt.
|
Tabelle 2: Studiendesign und Baseline-Merkmale der Studien 1 und 2 | ||
|
Studie 1 |
Studie 2 | |
|
Studientitel |
CAMMS323 (CARE-MS I) |
CAMMS32400507 (CARE-MS II) |
|
Studiendesign | ||
|
Anamnese |
Patienten mit aktiver MS, definiert als mindestens 2 Schübe in den vorangegangenen 2 Jahren. | |
|
Nachbeobachtung |
2 Jahre | |
|
Studienpopulation |
behandlungsnaive Patienten |
Patienten mit unzureichendem Ansprechen auf die vorherige Therapie* |
|
Baseline-Merkmale | ||
|
Mittleres Alter (Jahre) |
33 |
35 |
|
Mittlere/Mediane Krankheitsdauer |
2/1,6 Jahre |
4,5/3,8 Jahre |
|
Mittlere Dauer der vorherigen MS-Therapie (> 1 Arzneimittel angewendet) |
keine |
36 Monate |
|
% erhielten > 2 vorherige MS-Therapien |
Nicht zutreffend |
28% |
|
Mittlerer EDSS-Wert zur Baseline |
2,0 |
2,7 |
* Definiert als Patienten, die mindestens 1 Schub während der Behandlung mit beta-Interferon und Glatirameracetat erlitten, nachdem sie mindestens 6 Monate eine Behandlung mit dem Arzneimittel erhalten hatten
|
Tabelle 3: Wichtige klinische und MRT-Endpunkte aus den Studien 1 und 2 | ||||
|
Studie 1 |
Studie 2 | |||
|
Studientitel |
CAMMS323 (CARE-MS I) |
CAMMS32400507 (CARE-MS II) | ||
|
Klinische Endpunkte |
LEMTRADA 12 mg (N=376) |
IFNB-1a s.c. (N=187) |
LEMTRADA 12 mg (N=426) |
IFNB-1a s.c. (N=202) |
|
Schubrate1 Jährliche Schubrate (ARR) (95% KI) |
0,18 (0,13; 0,23) |
0,39 (0,29; 0,53) |
0,26 (0,21; 0,33) |
0,52 (0,41; 0,66) |
|
Inzidenzdichteratio (95% KI) Risikosenkung |
0,45 (0,32; 0,63) 54,9 (p<0,0001) |
0,51 (0,39; 0,65) 49,4 (p<0,0001) | ||
|
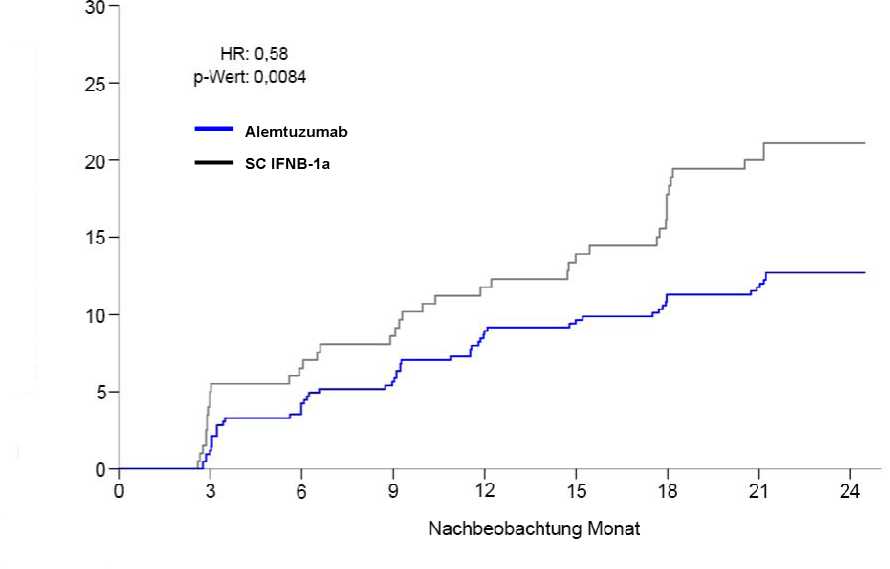
Behinderungen2 (Akkumulation von Behinderungen [SAD] über 6 Monate bestätigt1) Patienten mit SAD über 6 Monate bestätigt (95% KI) |
8,0% (5,7; 11,2) |
11,1% (7,3; 16,7) |
12,7% (9,9; 16,3) |
21,1% (15,9; 27,7) |
|
Risikoverhältnis (Hazard Ratio; 95% KI) |
0,70 (0,40; 1,23) (p=0,22) |
0,58 (0,38; 0,87) (p=0,0084) | ||
|
Schubfreie Patienten nach 2 Jahren (95% KI) |
77,6% (72,9; 81,6) (p<0,0001) |
58,7% (51,1; 65,5) |
65,4% (60,6; 69,7) (p<0,0001) |
46,7% (39,5; 53,5) |
|
Veränderung des EDSS ab Baseline nach 2 Jahren Schätzung (95% KI) |
-0,14 (-0,25; -0,02) (p=0,42) |
-0,14 (-0,29; 0,01) |
-0,17 (-0,29; -0,05) (p<0,0001) |
0,24 (0,07; 0,41) |
|
MRT-Endpunkte (0-2 Jahre) | ||||
|
Mediane prozentuale Veränderung des T2-Läsionsvolumens |
-9,3 (-19,6; -0,2) (p=0,31) |
-6,5 (-20,7; 2,5) |
-1,3 (p=0,14) |
-1,2 |
|
Patienten mit neuen oder sich vergrößernden T2-Läsionen über 2 Jahre |
48,5% (p=0,035) |
57,6% |
46,2% (p<0,0001) |
67,9% |
|
Patienten mit Gadolinium-anreichernde Läsionen über 2 Jahre |
15,4% (p=0,001) |
27,0% |
18,5% (p<0,0001) |
34,2% |
|
Patienten mit neuen T1-hypointensen Läsionen über 2 Jahre |
24,0% (p=0,055) |
31,4% |
19,9% (p<0,0001) |
38,0% |
|
Mediane prozentuale Veränderungen der Hirnathrophie |
-0,867 (p<0,0001) |
-1,488 |
-0,615 (p=0,012) |
-0,810 |
|
1 Koprimäre Endpunkte: ARR und SAD. Die Studie wurde als erfolgreich bewertet, wenn wenigstens einer der zwei koprimären Endpunkte erreicht wurde. 2 Die Zeit bis zum Auftreten von SAD war definiert als Anstieg um mindestens 1 Punkt auf der erweiterten Kurtzke-Skala (Expanded Disability Status Scale, EDSS), ausgehend von einem Baseline-EDSS-Wert von > 1,0 (Anstieg um 1,5 Punkte bei Patienten mit einem Baseline-EDSS-Wert von 0), über 6 Monate anhaltend. | ||||
Abbildung 1: Zeit bis zur 6 Monate anhaltenden Akkumulation von Behinderungen (Susta ined Accumulation of Disability, SAD) in Studie 2
Prozentualer Anteil an Patienten mit SAD
Schwere des Schubs
In Übereinstimmung mit der Wirkung auf die Schubrate zeigten unterstützende Analysen aus Studie 1 (CAMMS323), dass mit LEMTRADA 12 mg/Tag behandelte Patienten im Vergleich zu IFNB-1a signifikant weniger schwere Schübe erlitten (Rückgang um 61%, p=0,0056) und signifikant weniger Schübe, die mit Steroiden behandelt wurden (Rückgang um 58%, p<0,0001).
Unterstützende Analysen aus Studie 2 (CAMMS32400507) zeigten, das mit LEMTRADA 12 mg/Tag behandelte Patienten im Vergleich zu IFNB-1a signifikant weniger schwere Schübe erlitten (Rückgang um 48%, p=0,0121) und signifikant weniger Schübe, die mit Steroiden behandelt wurden (Rückgang um 56%, p<0,0001) oder einer Einweisung ins Krankenhaus bedurften (Rückgang um 55%, p<0,0045).
Anhaltender Rückgang von Behinderungen (Sustained Reduction of Disability, SRD)
Die Zeit bis zum Einsetzen von SRD war definiert als Rückgang um mindestens einen Punkt auf der EDSS ausgehend von einem Baseline-EDSS-Wert von > 2, der über mindestens 6 Monate anhielt. SRD ist eine Messgröße für eine anhaltende Verbesserung von Behinderungen. Insgesamt 29% der mit LEMTRADA behandelten Patienten erreichten einen SRD in Studie 2, während nur 13% der mit subkutan verabreichtem IFNB-1a behandelten Patienten diesen Endpunkt erreichten. Der Unterschied war statistisch signifikant
(p=0,0002).
Studie 3 (Phase-II-Studie CAMMS223) untersuchte über einen Zeitraum von 5 Jahren die Sicherheit und Wirksamkeit von LEMTRADA bei Patienten mit RRMS. Die Patienten wiesen einen EDSS-Wert von 0 - 3,0 auf, hatten in den vorangegangenen 2 Jahren mindestens 2 klinische Schübe und zeigten > 1 Gadolinium-angereicherte Läsion bei Eintritt in die Studie. Die Patienten hatten zuvor keine MS-Therapie erhalten. Die Patienten wurden mit LEMTRADA 12 mg/Tag (N=108) oder 24 mg/Tag (N=108), das in Monat 0 einmal täglich 5 Tage lang und in Monat 12 einmal täglich 3 Tage lang verabreicht wurde, oder mit 44 gg subkutan verabreichtem IFNB-1a (N=107), das 3 Jahre lang 3 mal pro Woche verabreicht wurde, behandelt. Sechsundvierzig Patienten erhielten in Monat 24 eine dritte Behandlungsphase mit LEMTRADA in einer Dosis von 12 mg/Tag oder 24 mg/Tag über 3 Tage.
Nach 3 Jahren senkte LEMTRADA das Risiko einer 6-monatigen SAD um 76% (Hazard Ratio: 0,24 [95%-KI: 0,110; 0,545], p<0,0006) und reduzierte die ARR um 67% (Inzidenzdichteratio: 0,33 [95%-KI: 0,196; 0,552], p<0,0001) im Vergleich zu subkutan verabreichtem IFNB-1a. Alemtuzumab 12 mg/Tag führte im Laufe der 2 Jahre Nachbeobachtung im Vergleich zu subkutan verabreichtem IFNB-1a zu signifikant niedrigeren EDSS-Werten (verbessert im Vergleich zur Baseline) (p<0,0001).
Nach 5 Jahren senkte LEMTRADA das Risiko einer SAD um 69% (Hazard Ratio: 0,31 [95%-KI: 0,161; 0,598], p=0,0005) und reduzierte die ARR um 66% (Inzidenzdichteratio: 0,34 [95%-KI: 0,202; 0,569], p<0,0001) im Vergleich zu subkutan verabreichtem IFNB-1a. .
In einer unverblindeten klinischen Nachbeobachtungsstudie zu LEMTRADA erhielten einige Patienten nach dokumentiertem Nachweis erneuter Krankheitsaktivität eine zusätzliche Behandlung mit LEMTRADA „nach Bedarf“. Die zusätzliche Behandlungsphase/die zusätzlichen Behandlungsphasen mit LEMTRADA wurde(n) in Dosen von 12 mg/Tag an 3 aufeinander folgenden Tagen (36 mg Gesamtdosis) im Abstand von mindestens 12 Monaten nach der vorherigen Behandlungsphase verabreicht. Der Nutzen und die Risiken von > 2 Behandlungsphasen sind nicht vollständig untersucht, aber die Ergebnisse weisen darauf hin, dass sich das Sicherheitsprofil bei zusätzlichen Behandlungsphasen nicht zu verändern scheint. Der Zeitraum bis zu einer eventuellen nächsten Behandlungsphase muss mindestens 12 Monate betragen.
Immunogenität
Wie bei allen therapeutischen Proteinen besteht ein Potential für Immunogenität. Die Daten reflektieren den prozentualen Anteil an Patienten, die unter Verwendung von ELISA (Enzyme-linked Immunosorbent Assay) positiv auf Antikörper gegen Alemtuzumab getestet wurden, was durch einen kompetitiven Bindungsassay bestätigt wurde. Positive Proben wurden mithilfe eines Durchflusszytometrie-Assays weiter auf Hinweise auf in-vitro-Hemmung untersucht. Von den Patienten in kontrollierten klinischen Studien zu MS wurden 1, 3 und 12 Monate nach jeder Behandlungsphase Serum-Proben zur Bestimmung von Anti-Alemtuzumab-Antikörpern genommen. Etwa 85% der Patienten, die LEMTRADA erhielten, wiesen während der Studie ein positives Testergebnis für Anti-Alemtuzumab-Antikörper auf, wobei 92% dieser Patienten auch ein positives Testergebnis für Antikörper zeigten, die die in-vitro-Bindung von LEMTRADA hemmten. Die Patienten, die Anti-Alemtuzumab-Antikörper aufwiesen, entwickelten diese 15 Monate nach der ersten Exposition. Es bestand kein Zusammenhang zwischen dem Vorliegen von Anti-Alemtuzumab-Antikörpern oder hemmenden Anti-Alemtuzumab-Antikörpern und einer Verminderung der Wirksamkeit, einer Veränderung bei der Pharmakokinetik oder dem Auftreten von Nebenwirkungen, einschließlich infusionsassoziierter Reaktionen.
Die Inzidenz von Antikörpern hängt stark von der Sensitivität und Spezifität des Assays ab. Darüber hinaus kann die beobachtete Inzidenz positiver Testergebnisse für Antikörper (einschließlich hemmender Antikörper) in einem Assay durch verschiedene Faktoren beeinflusst werden, einschließlich der Methode des Assays, Umgang mit den Proben, Zeitpunkt der Probenentnahme, gleichzeitig verabreichte Arzneimittel und vorbestehende Erkrankungen. Aus diesen Gründen kann der Vergleich der Inzidenz von Antikörpern gegen LEMTRADA mit der Inzidenz von Antikörpern gegen andere Arzneimittel irreführend sein.
Kinder und Jugendliche
Die Europäische Arzneimittel-Agentur hat für LEMTRADA eine Freistellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien über die Anwendung von Alemtuzumab bei Kindern im Alter von der Geburt bis zu unter zehn Jahren zur Behandlung von MS gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen).
Die Europäische Arzneimittel-Agentur hat für LEMTRADA eine Zurückstellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in einer oder mehreren pädiatrischen Altersklassen bei der Behandlung von schubförmig-remittierender MS (RRMS) gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen).
5.2 Pharmakokinetische Eigenschaften
Die Pharmakokinetik von LEMTRADA wurde bei insgesamt 216 Patienten mit schubförmig-remittierender MS (RRMS) untersucht, die intravenöse Infusionen von entweder 12 mg/Tag oder 24 mg/Tag an 5 aufeinander folgenden Tagen und 12 Monate nach der ersten Behandlungsphase an 3 aufeinander folgenden Tagen erhielten. Die Serumspiegel stiegen mit jeder weiteren Dosis innerhalb einer Behandlungsphase an, wobei die höchsten Spiegel nach der letzten Infusion einer Behandlungsphase beobachtet wurden. Die Verabreichung von 12 mg/Tag führte zu einer mittleren Cmax von 3014 ng/ml an Tag 5 der ersten Behandlungsphase und von 2276 ng/ml an Tag 3 der zweiten Behandlungsphase. Die Alpha-Halbwertszeit betrug etwa 4-5 Tage und war unter den Phasen vergleichbar, wobei innerhalb von 30 Tagen nach jeder Behandlungsphase niedrige oder nicht nachweisbare Serumspiegel erreicht wurden.
Alemtuzumab ist ein Protein, dessen erwarteter Stoffwechselweg der Abbau in kleine Peptide und einzelne Aminosäuren durch weit verteilte proteolytische Enzyme ist. Klassische Studien zur Biotransformation wurden nicht durchgeführt.
Schlussfolgerungen bezüglich der Wirkung von Ethnizität oder Geschlecht auf die Pharmakokinetik von LEMTRADA sind anhand der verfügbaren Daten nicht möglich. Die Pharmakokinetik von LEMTRADA bei Patienten im Alter über 55 Jahre wurde nicht untersucht.
5.3 Präklinische Daten zur Sicherheit
Karzinogenese und Mutagenese
Es wurden keine Studien zur Beurteilung des karzinogenen oder mutagenen Potentials von Alemtuzumab durchgeführt.
Fertilität, Schwangerschaft und Stillzeit
Die intravenöse Behandlung mit Alemtuzumab in Dosen von bis zu 10 mg/kg/Tag verabreicht an 5 aufeinander folgenden Tagen (AUC 7,1 mal höher als bei der menschlichen Exposition der empfohlenen täglichen Dosis) hatte keine Wirkung auf die Fertilität und Fruchtbarkeitsleistung männlicher transgener huCD52-Mäuse. Die Anzahl normaler Spermien war im Vergleich zu den Kontrollen signifikant reduziert (< 10%) und der prozentuale Anteil anomaler Spermien (schwanz- oder kopflos) war signifikant erhöht (bis zu 3%). Allerdings beeinträchtigten diese Veränderungen die Fertilität nicht und galten daher als unschädlich.
Bei weiblichen Mäusen, die vor der Kohabitation mit männlichen Mäusen des Wildtyps intravenös verabreichte Dosen von Alemtuzumab von bis zu 10 mg/kg/Tag (AUC 4,7 mal höher als bei der menschlichen Exposition der empfohlenen täglichen Dosis) verabreicht an 5 aufeinander folgenden Tagen erhielten, war die durchschnittliche Anzahl an Gelbkörpern und Implantationen pro Maus im Vergleich zu Vehikel-behandelten Tieren signifikant reduziert. Eine verringerte Gewichtszunahme während der Gestation im Vergleich zu Vehikel-behandelten Kontrollen wurde bei trächtigen Mäusen, die Dosen von 10 mg/kg/Tag erhielten, beobachtet.
Eine Studie zur Reproduktionstoxizität bei trächtigen Mäusen, die an 5 aufeinander folgenden Tagen während der Gestation intravenös verabreichte Dosen von Alemtuzumab von bis zu 10 mg/kg/Tag (AUC 2,4 Mal höher als bei der menschlichen Exposition der empfohlenen täglichen Dosis von 12 mg/kg/Tag) erhielten, zeigte einen signifikanten Anstieg der Anzahl von Muttertieren, bei denen alle Fruchtanlagen abstarben oder absorbiert wurden, sowie einen gleichzeitigen Rückgang bei der Anzahl an Muttertieren mit lebensfähigen Föten. Es wurden bei Dosen von bis zu 10 mg/kg/Tag keine externen, Weichteil- oder Skelettmissbildungen oder -veränderungen beobachtet.
Es wurden ein Plazentatransfer und eine potentielle pharmakologische Wirkung von Alemtuzumab bei Mäusen während der Gestation und nach der Geburt beobachtet. In Studien mit Mäusen wurden Veränderungen in den Lymphozytenzahlen bei Jungen beobachtet, die während der Gestation Alemtuzumab in Dosen von 3 mg/kg/Tag verabreicht an 5 aufeinander folgenden Tagen (AUC 0,6 Mal höher als bei der menschlichen Exposition der empfohlenen täglichen Dosis von 12 mg/Tag) ausgesetzt waren. Die kognitive, körperliche und sexuelle Entwicklung der Jungen, die durch das Säugen Alemtuzumab ausgesetzt waren, war bei Dosen von bis zu 10 mg/kg/Tag Alemtuzumab nicht beeinträchtigt.
6. PHARMAZEUTISCHE ANGABEN
6.1 Liste der sonstigen Bestandteile
Dinatriumphosphat-Dihydrat (E339)
Dinatriumedetat-Dihydrat Kaliumchlorid (E508)
Kaliumdihydrogenphosphat (E340)
Polysorbat 80 (E433)
Natriumchlorid
Wasser für Injektionszwecke
6.2 Inkompatibilitäten
Da keine Kompatibilitätsstudien durchgeführt wurden, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden, außer mit jenen, die in Abschnitt 6.6 erwähnt werden.
6.3 Dauer der Haltbarkeit
Konzentrat 3 Jahre
Verdünnte Lösung
Die chemische und physikalische Gebrauchsstabilität wurde bei 2°C bis 8°C über einen Zeitraum von 8 Stunden nachgewiesen.
Aus mikrobiologischer Perspektive sollte das Arzneimittel unmittelbar nach der Zubereitung verwendet werden. Bei einer nicht sofortigen Anwendung ist der Anwender für die Einhaltung einer entsprechenden Lagerungsdauer und entsprechender Lagerbedingungen unter Schutz vor Lichteinwirkung verantwortlich (nicht länger als 8 Stunden bei 2°C bis 8°C).
6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Konzentrat.
Im Kühlschrank lagern (2°C - 8°C).
Nicht einfrieren.
Durchstechflasche im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen. Aufbewahrungsbedingungen nach Verdünnung des Arzneimittels siehe Abschnitt 6.3.
6.5 Art und Inhalt des Behältnisses
LEMTRADA ist in einer durchsichtigen 2-ml-Durchstechflasche aus Glas mit Butylgummistopfen und Aluminiumsiegel mit einem Plastik-Schnappdeckel erhältlich.
Packungsgröße: Karton mit 1 Durchstechflasche.
6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung
Den Inhalt der Durchstechflasche vor der Anwendung auf Partikel und Verfärbungen überprüfen. Nicht verwenden, wenn Partikel vorhanden sind oder sich das Konzentrat verfärbt hat.
Die Durchstechflaschen vor Anwendung nicht schütteln.
Zur intravenösen Verabreichung nehmen Sie unter Anwendung aseptischer Methoden 1,2 ml LEMTRADA aus der Durchstechflasche in eine Spritze auf. Geben Sie es in 100 ml 0,9%ige NatriumchloridInfusionslösung (9 mg/ml) oder 5%ige Glukose-Infusionslösung. Dieses Arzneimittel darf nicht mit anderen Lösungsmitteln verdünnt werden. Der Beutel sollte vorsichtig geschwenkt werden, um die Lösung zu mischen.
LEMTRADA enthält keine antimikrobiellen Konservierungsmittel. Daher ist Vorsicht geboten, um die Sterilität der zubereiteten Lösung sicherzustellen. Es wird empfohlen, das verdünnte Arzneimittel unmittelbar zu verabreichen. Die Durchstechflaschen sind nur für den Einmalgebrauch bestimmt.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
7. INHABER DER ZULASSUNG
Genzyme Therapeutics Ltd 4620 Kingsgate Cascade Way
Oxford Business Park South
Oxford
OX4 2SU
Vereinigtes Königreich
8. ZULASSUNGSNUMMER(N)
EU/1/13/869/001
9. DATUM DER ERTEILUNG DER ZULASSUNG/VERLÄNGERUNG DER ZULASSUNG
Datum der Erteilung der Zulassung: 12. September 2013
10. STAND DER INFORMATION
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur http://www.ema.europa.eu/ verfügbar.
A. HERSTELLER DES WIRKSTOFFS BIOLOGISCHEN URSPRUNGS UND HERSTELLER, DIE FÜR DIE CHARGENFREIGABE VERANTWORTLICH SIND
B. BEDINGUNGEN ODER EINSCHRÄNKUNGEN FÜR DIE ABGABE UND DEN GEBRAUCH
C. SONSTIGE BEDINGUNGEN UND AUFLAGEN DER GENEHMIGUNG FÜR DAS INVERKEHRBRINGEN
D. BEDINGUNGEN ODER EINSCHRÄNKUNGEN FÜR DIE SICHERE UND WIRKSAME ANWENDUNG DES ARZNEIMITTELS
A. HERSTELLER DES WIRKSTOFFS BIOLOGISCHEN URSPRUNGS UND HERSTELLER, DIE FÜR DIE CHARGENFREIGABE VERANTWORTLICH SIND
Name und Anschrift des Herstellers des Wirkstoffs biologischen Ursprungs
Boehringer Ingelheim Pharma GmbH & Co. KG Birkendorfer Straße 65 88397 Biberach an der Riss Deutschland
Name und Anschrift der Hersteller, die für die Chargenfreigabe verantwortlich sind
Genzyme Limited 37 Hollands Road Haverhill Suffolk CB9 8PU
Vereinigtes Königreich
Genzyme Ireland Limited IDA Industrial Park Old Kilmeaden Road Waterford Irland
In der Druckversion der Packungsbeilage des Arzneimittels müssen Name und Anschrift des Herstellers, der für die Freigabe der betreffenden Charge verantwortlich ist, angegeben werden.
B. BEDINGUNGEN ODER EINSCHRÄNKUNGEN FÜR DIE ABGABE UND DEN GEBRAUCH
Arzneimittel auf eingeschränkte ärztliche Verschreibung (siehe Anhang I: Zusammenfassung der Merkmale des Arzneimittels, Abschnitt 4.2).
C. SONSTIGE BEDINGUNGEN UND AUFLAGEN DER GENEHMIGUNG FÜR DAS INVERKEHRBRINGEN
• Regelmäßig aktualisierte Unbedenklichkeitsberichte
Der Inhaber der Genehmigung für das Inverkehrbringen legt den ersten der regelmäßig zu aktualisierenden Unbedenklichkeitsberichte für dieses Arzneimittel innerhalb von 6 Monaten nach der Zulassung vor. Anschließend legt er regelmäßig aktualisierte Unbedenklichkeitsberichte für dieses Arzneimittel gemäß den Anforderungen der - nach Artikel 107 c Absatz 7 der Richtlinie 2001/83/EG vorgesehenen und im europäischen Internetportal für Arzneimittel veröffentlichten - Liste der in der Union festgelegten Stichtage (EURD-Liste) vor.
D. BEDINGUNGEN ODER EINSCHRÄNKUNGEN FÜR DIE SICHERE UND WIRKSAME ANWENDUNG DES ARZNEIMITTELS
• Risikomanagement-Plan (RMP)
Der Inhaber der Genehmigung für das Inverkehrbringen führt die notwendigen, im vereinbarten RMP beschriebenen und in Modul 1.8.2 der Zulassung dargelegten Pharmakovigilanzaktivitäten und
Maßnahmen sowie alle künftigen vom Ausschuss für Humanarzneimittel (CHMP) vereinbarten Aktualisierungen des RMP durch.
Ein aktualisierter RMP ist einzureichen:
• nach Aufforderung durch die Europäische Arzneimittel-Agentur;
• jedes Mal wenn das Risikomanagement-System geändert wird, insbesondere infolge neuer eingegangener Informationen, die zu einer wesentlichen Änderung des Nutzen-Risiko-Verhältnisses führen können oder infolge des Erreichens eines wichtigen Meilensteins (in Bezug auf Pharmakovigilanz oder Risikominimierung).
Fallen die Vorlage eines PSUR und die Aktualisierung eines RMP zeitlich zusammen, können beide gleichzeitig vorgelegt werden.
• Zusätzliche Maßnahmen zur Risikominimierung
Vor dem Inverkehrbringen in jedem EU-Mitgliedsstaat muss der Inhaber der Genehmigung für das Inverkehrbringen ein Fortbildungsprogramm für medizinische Fachkreise und Patienten mit der national zuständigen Behörde abstimmen.
Der Inhaber der Genehmigung für das Inverkehrbringen soll sicherstellen, dass nach der Vereinbarung mit der national zuständigen Behörde in jedem Mitgliedsstaat, in dem LEMTRADA verkauft wird, alle Ärzte, die dieses Medikament zu verschreiben beabsichtigen, zur und nach der Markteinführung ein aktuelles Fortbildungspaket für Ärzte erhalten, das aus Folgendem besteht:
• der Fachinformation
• dem Leitfaden für Ärzte
• einer Checkliste für den verschreibenden Arzt
• dem Leitfaden für Patienten
• der Patientenkarte
Der Leitfaden für Ärzte muss folgende wichtige Hinweise enthalten:
1. eine Beschreibung der mit der Einnahme von LEMTRADA verbundenen Risiken, nämlich:
• Idiopathische thrombozytopenische Purpura (ITP)
• Nephropathien, einschließlich Goodpasture-Syndrom (anti-GBM-Krankheit)
• Schilddrüsenerkrankungen
2. Empfehlungen zur Risikominimierung durch geeignete Beratung, Kontrolle und Management der Patienten
3. einen Abschnitt "Häufig gestellte Fragen"
Die Checkliste für den verschreibenden Arzt soll folgende wichtige Punkte umfassen:
1. Auflistung von Tests, die bei der ersten Untersuchung des Patienten durchgeführt werden müssen
2. Hinweis auf die Notwendigkeit abgeschossener Impfungen (erste Impfstoffverabreichung und Auffrischungsimpfung) 6 Wochen vor Behandlungsbeginn.
3. Überprüfung der Möglichkeit einer Prämedikation, des allgemeinen Gesundheitszustands sowie die Durchführung eines Schwangerschaftstests unmittelbar vor Behandlungsbeginn
4. Kontrolluntersuchungen während der Behandlung und vier Jahre lang nach der letzten Behandlung
5. einen spezielle Verweis darauf, dass der Patient über die Risiken schwerer
Autoimmunerkrankungen, Infektionen und bösartige Neubildungen, sowie die Maßnahmen zur Risikominimierung informiert wurde und sie verstanden hat
Der Leitfaden für Patienten soll folgende wichtige Hinweise enthalten:
1. eine Beschreibung der mit der Einnahme von LEMTRADA verbundenen Risiken, nämlich:
• Idiopathische thrombozytopenische Purpura (ITP)
• Nephropathien, einschließlich Goodpasture-Syndrom (anti-GBM-Krankheit)
• Schilddrüsenerkrankungen
• schwere Infektionen
2. eine Beschreibung der Anzeichen und Symptome von Autoimmunerkrankungen
3. eine Beschreibung der besten Vorgehensweise, wenn Anzeichen und entsprechende Symptome für Autoimmunkrankheiten auftreten (z. B., wie Patienten Ihren Arzt erreichen können)
4. Empfehlungen für die Planung der Kontrolluntersuchungen Die Patientenkarte soll folgende wichtige Informationen enthalten:
1. Einen Hinweis an alle Ärzte, die den Patienten zu einem beliebigen Zeitpunkt und auch in Notfällen behandeln, dass dieser mit LEMTRADA behandelt wurde.
2. dass LEMTRADA das Risiko erhöhen kann, folgende Krankheiten zu bekommen:
• Idiopathische thrombozytopenische Purpura (ITP)
• Nephropathien, einschließlich Goodpasture-Syndrom (anti-GBM-Erkrankung)
• Schilddrüsenerkrankungen
• schwere Infektionen
3. die Kontaktdaten des Arztes, der LEMTRADA verschrieben hat
ETIKETTIERUNG UND PACKUNGSBEILAGE
A. ETIKETTIERUNG
LEMTRADA 12 mg Konzentrat zur Herstellung einer Infusionslösung Alemtuzumab
Jede Durchstechflasche enthält 12 mg Alemtuzumab in 1,2 ml (10 mg/ml).
E339, Dinatriumedetat-Dihydrat, E508, E340, E433, Natriumchlorid, Wasser für Injektionszwecke
Konzentrat zur Herstellung einer Infusionslösung 1 Durchstechflasche 12 mg/1,2 ml
Packungsbeilage beachten.
Intravenöse Anwendung.
Innerhalb von 8 Stunden nach der Verdünnung verabreichen.
Arzneimittel für Kinder unzugänglich aufbewahren.
Verwendbar bis:
Die Durchstechflasche im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Im Kühlschrank lagern.
Nicht einfrieren-oder schütteln.
10. GEGEBENENFALLS BESONDERE VORSICHTSMASSNAHMEN FÜR DIE BESEITIGUNG VON NICHT VERWENDETEM ARZNEIMITTEL ODER DAVON STAMMENDEN ABFALLMATERIALIEN
Genzyme Therapeutics Ltd 4620 Kingsgate Cascade Way
Oxford Business Park South
Oxford
OX4 2SU
Vereinigtes Königreich
EU/1/13/869/001
Ch.-B.:
<Der Begründung, keine Angaben in Blindenschrift aufzunehmen, wird zugestimmt.>
MINDESTANGABEN AUF KLEINEN BEHÄLTNISSEN ETIKETT/DURCHSTECHFLASCHE
1. BEZEICHNUNG DES ARZNEIMITTELS SOWIE ART(EN) DER ANWENDUNG
LEMTRADA 12 mg steriles Konzentrat
Alemtuzumab
i.v.
2. HINWEISE ZUR ANWENDUNG
3. VERFALLDATUM
EXP
4. CHARGENBEZEICHNUNG
Lot
5. INHALT NACH GEWICHT, VOLUMEN ODER EINHEITEN
1,2 ml
6. WEITERE ANGABEN
B. PACKUNGSBEILAGE
Gebrauchsinformation: Information für Patienten
LEMTRADA 12 mg Konzentrat zur Herstellung einer Infusionslösung
Alemtuzumab
Dieses Arzneimittel miterliegt einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Sie können dabei helfen, indem Sie jede auftretende Nebenwirkung melden. Hinweise zur Meldung von Nebenwirkungen, siehe Ende Abschnitt 4.
Lesen Sie die gesamte Packungsbeilage sorgfältig durch, bevor Ihnen dieses Arzneimittel verabreicht wird, denn sie enthält wichtige Informationen.
- Heben Sie diese Packungsbeilage auf. Vielleicht möchten Sie diese später nochmals lesen.
- Wenn Sie weitere Fragen haben, wenden Sie sich an Ihren Arzt.
- Wenn Sie Nebenwirkungen bemerken, wenden Sie sich an Ihren Arzt. Dies gilt auch für Nebenwirkungen, die nicht in dieser Packungsbeilage angegeben sind. Siehe Abschnitt 4.
Was in dieser Packungsbeilage steht
1. Was ist LEMTRADA und wofür wird es angewendet?
2. Was sollten Sie vor der Verabreichung von LEMTRADA beachten?
3. Wie wird LEMTRADA verabreicht?
4. Welche Nebenwirkungen sind möglich?
5. Wie ist LEMTRADA aufzubewahren?
6. Inhalt der Packung und weitere Informationen
1. Was ist LEMTRADA und wofür wird es angewendet?
LEMTRADA enthält den Wirkstoff Alemtuzumab, der zur Behandlung einer bestimmten Form der Multiplen Sklerose (MS) bei Erwachsenen, die als schubförmig-remittierende Multiple Sklerose (RRMS) bezeichnet wird, angewendet wird. LEMTRADA heilt MS nicht, kann aber die Anzahl an Schüben der MS senken. Es kann auch dabei helfen, dass sich einige der Zeichen und Symptome der MS verlangsamen oder zurückbilden. In klinischen Studien hatten mit LEMTRADA behandelte Patienten weniger Schübe und eine geringere Wahrscheinlichkeit einer Verschlechterung der Behinderung als Patienten, denen mehrmals pro Woche beta-Interferon injiziert wurde.
Was ist Multiple Sklerose?
MS ist eine Autoimmunerkrankung, die das Zentralnervensystem (Gehirn und Rückenmark) befällt. Bei MS greift Ihr Immunsystem fälschlicherweise die Schutzschicht (Myelin) rund um die Nervenstränge an, was zu einer Entzündung führt. Wenn die Entzündung Symptome hervorruft, wird dies oft als „Schub“ bezeichnet. Bei RRMS erleiden die Patienten Schübe, gefolgt von symptomfreien Phasen.
Die Symptome, die Sie erleiden, werden davon bestimmt, welcher Teil des Zentralnervensystems betroffen ist. Der Schaden, den Ihre Nerven während der Entzündung nehmen, kann reversibel sein. Aber im weiteren Verlauf der Erkrankung kann der Schaden zunehmen und dauerhaft werden.
Wie wirkt LEMTRADA?
LEMTRADA reguliert Ihr Immunsystem, um seine Angriffe auf Ihr Zentralnervensystem zu begrenzen.
2. Was sollten Sie vor der Verabreichung von LEMTRADA beachten? LEMTRADA darf NICHT angewendet werden,
- wenn Sie allergisch gegen Alemtuzumab oder einen der in Abschnitt 6. genannten sonstigen Bestandteile dieses Arzneimittels sind
- wenn Sie mit dem Human-Immunodeficiency-Virus infiziert sind (HIV-Infektion).
Warnhinweise und Vorsichtsmaßnahmen
Bitte sprechen Sie vor der Verabreichung von LEMTRADA mit Ihrem Arzt. Nach einer Behandlungsphase mit LEMTRADA unterliegen Sie einem erhöhten Risiko, Autoimmunerkrankungen zu entwickeln oder schwere Infektionen zu erleiden. Es ist wichtig, dass Sie diese Risiken verstehen und wissen, wie Sie sich in Hinblick auf diese selbst beobachten. Es werden Ihnen eine Patientenkarte und ein Leitfaden für Patienten mit weiteren Informationen ausgehändigt. Es ist wichtig, dass Sie die Patientenkarte während der Behandlungsdauer und über 4 Jahre nach der letzten Infusion von LEMTRADA bei sich führen, da Nebenwirkungen auch noch mehrere Jahre nach der Behandlung auftreten können. Wenn Sie sich einer medizinischen Behandlung unterziehen, auch wenn es sich nicht um eine Behandlung der MS handelt, zeigen Sie dem jeweiligen Arzt Ihre Patientenkarte vor.
Ihr Arzt wird vor dem Beginn der Behandlung mit LEMTRADA Blutuntersuchungen durchführen. Diese Untersuchungen werden vorgenommen, um zu sehen, ob Ihnen LEMTRADA verabreicht werden kann. Ihr Arzt wird auch sicherstellen wollen, dass bei Ihnen bestimmte Erkrankungen oder Störungen ausgeschlossen werden können, bevor Sie die Behandlung mit LEMTRADA beginnen.
• Autoimmunerkrankungen
Die Behandlung mit LEMTRADA kann das Risiko für Autoimmunerkrankungen erhöhen. Dies sind Erkrankungen, bei denen Ihr Immunsystem fälschlicherweise Ihren Körper angreift. Informationen über einige spezifische Erkrankungen, die bei mit LEMTRADA behandelten MS-Patienten beobachtet wurden, sind im Folgenden aufgeführt.
Die Autoimmunerkrankungen können noch mehrere Jahre nach der Behandlung mit LEMTRADA auftreten. Daher sind bis 4 Jahre nach Ihrer letzten Infusion regelmäßige Blut- und Urinuntersuchungen erforderlich. Die Untersuchungen sind auch notwendig, wenn Sie sich gut fühlen und Ihre MS-Symptome unter Kontrolle sind. Darüber hinaus gibt es bestimmte Zeichen und Symptome, auf die Sie bei sich achten sollten. Näheres zu den Zeichen und Symptomen, den Untersuchungen und den zu ergreifenden Maßnahmen finden Sie in Abschnitt 4 - Autoimmunerkrankungen.
Hilfreichere Informationen über diese Autoimmunerkrankungen (und entsprechende Untersuchungen) finden Sie im LEMTRADA-Leitfaden für Patienten.
o Idiopathische thrombozytopenische Purpura (ITP)
Gelegentlich entwickelten Patienten eine Blutungsstörung aufgrund niedriger Blutplättchenspiegel, die als idiopathische thrombozytopenische Purpura (ITP) bezeichnet wird. Sie muss frühzeitig diagnostiziert und behandelt werden, da ihre Auswirkungen andernfalls schwerwiegend oder sogar tödlich sein können. Die Zeichen und Symptome von ITP sind unter Abschnitt 4 beschrieben.
o Nierenerkrankung (wie etwa anti-GBM-Krankheit)
Selten entwickelten Patienten Autoimmunerkrankungen der Nieren, wie z. B. das Goodpasture-Syndrom (anti-GBM-Krankheit). Die Zeichen und Symptome von Nierenerkrankungen sind unter Abschnitt 4 beschrieben. Eine nicht behandelte Nierenerkrankung kann zu Nierenversagen führen, was wiederum eine Dialyse oder eine Transplantation erfordert und zum Tod führen kann.
o Schilddrüsenerkrankungen
Sehr häufig entwickelten Patienten eine Autoimmunerkrankung der Schilddrüse, welche deren Fähigkeit zur Bildung oder Kontrolle von Hormonen, die für den Stoffwechsel wichtig sind, beeinträchtigte.
LEMTRADA kann verschiedene Formen von Schilddrüsenerkrankungen verursachen, einschließlich:
• eine Schilddrüsenüberfunktion (Hyperthyreose), wenn die Schilddrüse zu viele Hormone produziert
• eine Schilddrüsenunterfunktion (Hypothyreose), wenn die Schilddrüse nicht genug Hormone produziert.
Die Zeichen und Symptome von Schilddrüsenerkrankungen sind unter Abschnitt 4 beschrieben.
Wenn Sie eine Schilddrüsenerkrankung entwickeln, müssen Sie in den meisten Fällen für den Rest Ihres Lebens mit Arzneimitteln zur Kontrolle Ihrer Schilddrüsenerkrankung behandelt werden. In einigen Fällen muss die Schilddrüse entfernt werden.
Es ist sehr wichtig, dass Ihre Schilddrüsenerkrankung korrekt behandelt wird, insbesondere wenn Sie nach der Anwendung von LEMTRADA schwanger werden. Eine unbehandelte Schilddrüsenerkrankung kann Ihrem Kind vor oder nach der Geburt schaden.
o Andere Autoimmunerkrankungen
Selten entwickelten Patienten Autoimmunerkrankungen in Bezug auf die roten oder weißen Blutkörperchen. Diese können anhand der Blutuntersuchungen diagnostiziert werden, die bei Ihnen nach der Behandlung mit LEMTRADA regelmäßig durchgeführt werden. Wenn Sie eine dieser Erkrankungen entwickeln, wird Ihr Arzt Ihnen das mitteilen und entsprechende Maßnahmen ergreifen, um sie zu behandeln.
• Infusionsreaktionen
Die meisten mit LEMTRADA behandelten Patienten weisen Nebenwirkungen zum Zeitpunkt der Infusion oder innerhalb von 24 Stunden nach der Infusion auf. Ihr Arzt wird Ihnen (ein) andere(s) Arzneimittel geben, um zu versuchen, die Infusionsreaktionen zu reduzieren (siehe Abschnitt 4 - Infusionsreaktionen).
• Infektionen
Mit LEMTRADA behandelte Patienten haben ein höheres Risiko für schwerwiegende Infektionen (siehe Abschnitt 4 -Infektionen). Im Allgemeinen können die Infektionen mit gängigen Arzneimitteln behandelt werden.
Um die Wahrscheinlichkeit des Auftretens einer Infektion zu senken, wird Ihr Arzt überprüfen, ob andere Arzneimittel, die Sie einnehmen, Ihr Immunsystem beeinträchtigen könnten. Aus diesem Grund ist es wichtig, dass Sie Ihren Arzt über alle Arzneimittel informieren, die Sie einnehmen.
Wenn Sie eine Infektion haben, bevor Sie mit der Behandlung mit LEMTRADA beginnen, wird Ihr Arzt außerdem in Erwägung ziehen, die Behandlung zu verschieben, bis die Infektion unter Kontrolle oder vollständig zurückgebildet ist.
Mit LEMTRADA behandelte Patienten haben ein höheres Risiko eine Herpes-Infektion (z. B. oraler Herpes) zu bekommen. Im allgemeinen hat ein Patient, der einmal eine Herpes-Infektion hatte, ein höheres Risiko für ein erneutes Auftreten. Es kann auch sein, dass Sie zum ersten Mal Herpes bekommen. Es wird empfohlen, dass Ihr Arzt Ihnen Arzneimittel verschreibt, um die Wahrscheinlichkeit des Auftretens einer Herpes-Infektion zu senken. Diese Arzneimittel sollten an den Tagen, an denen Sie mit LEMTRADA behandelt werden, und einen Monat lang nach der Behandlung, eingenommen werden.
Darüber hinaus sind Infektionen möglich, die zu Anomalien der Zervix (Gebärmutterhals) führen können. Daher wird empfohlen, dass alle Patientinnen ein jährliches Screening, wie etwa einen Zervixabstrich, vornehmen lassen. Ihr Arzt wird Ihnen erklären, welche Untersuchungen Sie benötigen.
Patienten, die mit LEMTRADA behandelt wurden, haben auch ein höheres Risiko, eine Listerieninfektion zu bekommen (eine bakterielle Infektion, die durch Aufnahme kontaminierter Lebensmittel ausgelöst wird). Eine Listerieninfektion kann ernsthafte Erkrankungen, wie z.B. eine Meningitis (Hirnhautentzündung), auslösen. Um dieses Risiko zu reduzieren, sollten Sie kein rohes oder nicht durchgegartes Fleisch, keinen
Weichkäse und keine unpasteurisierten Milchprodukte bis mindestens einen Monat nach der LEMTRADA-Behandlung verzehren.
Wenn Sie in einer Region leben, in der Tuberkuloseinfektionen häufig vorkommen, unterliegen Sie eventuell einem erhöhten Risiko für eine Tuberkuloseinfektion. Tuberkulose-Untersuchungen werden von Ihrem Arzt vereinbart.
Wenn Sie Träger des Hepatitis-B- oder Hepatitis-C-Virus (welche sich auf die Leber auswirken) sind, ist vor Verabreichung von LEMTRADA besondere Vorsicht geboten, da unbekannt ist, ob die Behandlung zu einer Aktivierung der Hepatitis-Infektion führen kann, welche in der Folge Ihrer Leber schaden könnte.
• Früher diagnostizierte Krebserkrankung
Wenn bei Ihnen in der Vergangenheit eine Krebserkrankung festgestellt wurde, informieren Sie bitte Ihren Arzt darüber.
• Impfstoffe
Es ist nicht bekannt, ob LEMTRADA Ihr Ansprechen auf Impfstoffe beeinträchtigt. Wenn Sie die StandardImpfungen nicht vollständig erhalten haben, wird Ihr Arzt überlegen, ob Sie sie vor der Behandlung mit LEMTRADA erhalten sollten. Insbesondere wird Ihr Arzt in Erwägung ziehen, Sie gegen Windpocken zu impfen, wenn Sie diese noch nicht hatten. Alle Impfung müssen mindestens 6 Wochen vor Beginn einer Behandlungsphase mit LEMTRADA verabreicht werden.
Sie dürfen bestimmte Arten von Impfstoffen (Lebendimpfstoffe) NICHT erhalten, wenn Ihnen vor kurzem LEMTRADA verabreicht wurde.
Kinder und Jugendliche
LEMTRADA ist nicht für die Anwendung bei Kindern und Jugendlichen unter 18 Jahren vorgesehen, da es bei MS-Patienten unter 18 Jahren nicht untersucht wurde.
Einnahme von LEMTRADA zusammen mit anderen Arzneimitteln
Informieren Sie Ihren Arzt oder Apotheker, wenn Sie andere Arzneimittel anwenden, kürzlich andere Arzneimittel angewendet haben oder beabsichtigen, andere Arzneimittel anzuwenden (einschließlich Impfungen und pflanzlicher Arzneimittel).
Neben LEMTRADA gibt es andere Behandlungen (einschließlich Behandlungen von MS oder auch anderen Erkrankungen), die Ihr Immunsystem beeinflussen und somit Ihre Abwehr gegen Infektionen beeinträchtigen könnten. Wenn Sie eine solche Behandlung erhalten, kann Ihr Arzt Sie auffordern, die anderen Arzneimittel vor Beginn der Behandlung mit LEMTRADA abzusetzen.
Schwangerschaft
Wenn Sie schwanger sind, wenn Sie vermuten, schwanger zu sein oder beabsichtigen, schwanger zu werden, fragen Sie vor der Anwendung dieses Arzneimittels Ihren Arzt um Rat.
Frauen im gebärfähigen Alter sollten während und 4 Monate lang nach einer Behandlungsphase mit LEMTRADA eine zuverlässige Verhütungsmethode anwenden.
Wenn Sie nach der Anwendung von LEMTRADA schwanger werden und während der Schwangerschaft eine Schilddrüsenerkrankung entwickeln, ist besondere Vorsicht geboten. Schilddrüsenerkrankungen könnten dem Baby schaden (siehe Abschnitt 2 Warnhinweise und Vorsichtsmaßnahmen -Autoimmunerkrankungen).
Stillzeit
Es ist nicht bekannt, ob LEMTRADA über die Muttermilch auf ein Baby übertragen werden kann, aber es besteht die Möglichkeit, dass dies der Fall ist. Es wird empfohlen, dass Sie während und 4 Monate lang nach jeder Behandlungsphase mit LEMTRADA nicht stillen. Allerdings kann Muttermilch (die Ihr Baby vor
Infektionen schützen kann) Vorteile haben. Sprechen Sie daher mit Ihrem Arzt, wenn Sie planen, Ihr Baby zu stillen. Er wird mit Ihnen darüber sprechen, was für Sie und Ihr Baby das Richtige ist.
Zeugungs-/Gebärfähigkeit
Während der Behandlungsphase und 4 Monate lang danach kann sich LEMTRADA in Ihrem Körper befinden. Es ist nicht bekannt, ob LEMTRADA während dieser Zeit eine Wirkung auf die Zeugungs-/Gebärfähigkeit hat. Sprechen Sie mit Ihrem Arzt, wenn Sie darüber nachdenken, schwanger werden zu wollen.
Verkehrstüchtigkeit und Fähigkeit zum Bedienen von Maschinen
Bei zahlreichen Patienten treten während und bis zu 24 Stunden nach der Infusion von LEMTRADA Nebenwirkungen auf. Einige davon, zum Beispiel Schwindelgefühl, können die Verkehrstüchtigkeit und das Bedienen von Maschinen beeinträchtigen. Wenn dies auf Sie zutrifft, führen Sie diese Tätigkeiten nicht aus, bis Sie sich besser fühlen.
LEMTRADA enthält Kalium und Natrium.
Dieses Arzneimittel enthält weniger als 1 mmol Kalium (39 mg) pro Infusion, d. h. es ist im Grunde „kaliumfrei“.
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Infusion, d. h. es ist im Grunde „natriumfrei“.
3. Wie wird LEMTRADA verabreicht?
Ihr Arzt wird Ihnen erklären, wie LEMTRADA verabreicht wird. Wenn Sie Fragen haben, wenden Sie sich an Ihren Arzt.
Bei der ersten Behandlungsphase erhalten Sie an 5 aufeinanderfolgenden Tagen je eine Infusion pro Tag (Phase 1).
Ein Jahr später erhalten Sie an 3 aufeinanderfolgenden Tagen je eine Infusion pro Tag (Phase 2).
Zwischen den zwei Phasen findet keine Behandlung mit LEMTRADA statt.
Die tägliche Höchstdosis ist eine Infusion.
LEMTRADA wird Ihnen als Infusion in eine Vene verabreicht. Jede Infusion dauert etwa 4 Stunden. Bei den meisten Patienten senken 2 Behandlungsphasen die MS-Aktivität über 2 Jahre. Eine Überwachung in Hinblick auf Nebenwirkungen sowie regelmäßige Untersuchungen müssen bis 4 Jahre nach der letzten Infusion fortgeführt werden.
Um die Dauer der Wirkungen der Behandlung und die Dauer der erforderlichen Kontrolluntersuchungen
besser zu verstehen, sehen Sie sich bitte das folgende Schaubild an.
Erste Blut- und Urinuntersuchung vor der Behandlung
-►- und in den 4 Jahren nach der letzten Infusion
1. Behandlungsphase 2. Behandlungsphase
lllll III
5-tägige Behandlung 3-tägige Behandlung
Jahr 1 Jahr 2 Jahr 3 Jahr 4 Jahr 5
HINWEIS Bei den meisten Patienten wirken 2 Behandlungsphasen 2 Jahre o der länger.
BEHANDLUNG
KONTR OT ,T .UNTERSUCHUNGEN
Nachbeobachtung nach der Behandlung mit LEMTRADA
Nachdem Sie LEMTRADA erhalten haben, werden Sie sich regelmäßigen Untersuchungen unterziehen müssen, um sicherzustellen, dass jegliche potentielle Nebenwirkungen frühzeitig diagnostiziert und behandelt werden. Diese Untersuchungen müssen 4 Jahre lang nach der letzten Infusion fortgeführt werden und sind unter Abschnitt 4, die wichtigsten Nebenwirkungen, aufgeführt.
Wenn Sie eine größere Menge von LEMTRADA erhalten haben, als Sie sollten
Patienten, die bei einer Infusion versehentlich zu viel LEMTRADA erhalten hatten, entwickelten schwerwiegende Reaktionen, wie etwa Kopfschmerzen, Hautausschlag, niedriger Blutdruck oder erhöhte Herzfrequenz. Dosen über der empfohlenen Dosis können zu schwereren und länger anhaltenden Infusionsreaktionen führen (siehe Abschnitt 4) oder eine stärkere Wirkung auf das Immunsystem haben. Die Behandlung besteht darin, die Verabreichung von LEMTRADA abzubrechen und die Symptome zu behandeln.
Wenn Sie weitere Fragen zur Anwendung dieses Arzneimittels haben, wenden Sie sich an Ihren Arzt.
4. Welche Nebenwirkungen sind möglich?
Wie alle Arzneimittel kann LEMTRADA Nebenwirkungen haben, die aber nicht bei jedem auftreten müssen.
Die wichtigsten Nebenwirkungen sind die unter Abschnitt 2 beschriebenen
Autoimmunerkrankungen. Dazu zählen:
• ITP (Blutungsstörung) (gelegentlich - kann bei bis zu 1 von 100 Patienten auftreten): kann sich als vereinzelte, kleine rote, rosafarbene oder violette Punkte auf der Haut, Neigung zu Blutergüssen, schwierigeres Stillen von Blutungen bei Schnitten, stärkere, längere oder häufigere Menstruation als normal, Zwischenblutungen, Zahnfleisch- oder Nasenbluten, die neu aufgetreten sind oder länger als normal brauchen, bis sie gestillt sind, oder das Abhusten von Blut, äußern.
• Nierenerkrankungen (selten - können bei bis zu 1 von 1.000 Patienten auftreten): können sich als Blut im Urin (Ihr Urin kann rot oder teefarben sein) oder Anschwellen von Beinen oder Füßen äußern. Es können auch Lungenschädigungen auftreten, was zum Abhusten von Blut führen kann.
Wenn Sie eines dieser Zeichen oder Symptome von Blutungsstörungen oder Nierenerkrankungen bei sich feststellen, rufen Sie unverzüglich Ihren Arzt an, um ihm die Symptome zu berichten. Wenn Sie Ihren Arzt nicht erreichen können, suchen Sie umgehend anderweitige medizinische Versorgung auf.
• Schilddrüsenerkrankungen (sehr häufig - können bei mehr als 1 von 10 Patienten auftreten): können sich als übermäßiges Schwitzen, ungeklärte Gewichtsabnahme oder -zunahme, Schwellung des Auges, Nervosität, Herzrasen, Kältegefühl, zunehmende Müdigkeit oder neu aufgetretene Verstopfung äußern.
• Störungen der roten und weißen Blutkörperchen (selten - können bei bis zu 1 von 1.000 Patienten auftreten): diagnostiziert anhand Ihrer Blutuntersuchungen.
All diese schweren Nebenwirkungen können auch noch mehrere Jahre nach der Behandlung mit LEMTRADA auftreten. Wenn Sie eines dieser Zeichen oder Symptome bei sich feststellen, rufen Sie unverzüglich Ihren Arzt an, um ihm diese zu berichten. Sie werden sich auch regelmäßig Blut- und Urinuntersuchungen unterziehen müssen, um sicherzustellen, dass Sie im Falle eines Auftretens einer dieser Erkrankungen umgehend behandelt werden._
Zusammenfassung der Laboruntersuchungen, die bei Ihnen in Hinblick auf Autoimmunerkrankungen durchgeführt werden:
|
Untersuchung |
Wann? |
Wie lange? |
|
Blutuntersuchung (zur Diagnose aller o. g. wichtigen schwerwiegenden Nebenwirkungen) |
Vor Beginn der Behandlung und nach der Behandlung jeden Monat |
Bis 4 Jahre nach der letzten LEMTRADA-Infusion |
|
Urinuntersuchung (zusätzliche Untersuchung zur Diagnose von Nierenerkrankungen) |
Vor Beginn der Behandlung und nach der Behandlung jeden Monat |
Bis 4 Jahre nach der letzten LEMTRADA-Infusion |
Wenn bei Ihnen nach diesem Zeitraum Symptome einer ITP, Nieren- oder Schilddrüsenerkrankung auftreten, wird Ihr Arzt weitere Untersuchungen durchführen. Sie sollten auch nach 4 Jahren aufpassen, ob es Zeichen von Nebenwirkungen gibt und entsprechende Symptomen auftreten, wie in Ihrem Leitfaden für Patienten ausgeführt, und weiterhin die Patientenkarte bei sich tragen.
Eine weitere wichtige Nebenwirkung ist ein erhöhtes Risiko für Infektionen (weitere Informationen, wie oft Patienten Infektionen erleiden, siehe unten). In den meisten Fällen sind diese leicht, aber es können auch schwerwiegende Infektionen auftreten.
Informieren Sie bitte unverzüglich Ihren Arzt, wenn Sie eines dieser Zeichen einer Infektion bei sich feststellen.
• Fieber und/oder Schüttelfrost
_• geschwollene Lymphknoten_
Um zu helfen, das Risiko einiger Infektionen zu senken, kann Ihr Arzt in Erwägung ziehen, Ihnen eine Windpocken-Impfung und/oder andere Impfungen, die er bei Ihnen für erforderlich hält, zu verabreichen (siehe Abschnitt 2 - Was sollten Sie vor der Verabreichung von LEMTRADA beachten? - Impfstoffe). Ihr Arzt kann Ihnen auch ein Arzneimittel gegen oralen Herpes verschreiben (siehe Abschnitt 2 - Was sollten Sie vor der Verabreichung von LEMTRADA beachten? - Infektionen).
Die häufigsten Nebenwirkungen sind Infusionsreaktionen (weitere Informationen, wie oft Patienten diese erleiden, siehe unten), die zum Zeitpunkt der Infusion oder innerhalb von 24 Stunden nach der Infusion auftreten können. In den meisten Fällen sind diese leicht, aber es sind auch einige schwerwiegende Reaktionen möglich. Gelegentlich können allergische Reaktionen auftreten.
Um zu versuchen, die Infusionsreaktionen zu reduzieren, wird Ihr Arzt Ihnen ein Arzneimittel (Kortikosteroide) vor jeder der ersten 3 Infusionen einer LEMTRADA-Phase geben. Andere Behandlungen zur Begrenzung dieser Reaktionen können ebenfalls vor der Infusion oder bei Auftreten von Symptomen verabreicht werden. Darüber hinaus werden Sie während der Infusion und 2 Stunden lang nach Abschluss dieser beobachtet. Im Falle schwerwiegender Reaktionen wird die Infusion verlangsamt oder gar beendet.
Bitten lesen Sie den LEMTRADA-Leitfaden für Patienten für weitere Informationen über diese Nebenwirkungen.
Dies sind die Nebenwirkungen, die auftreten können.
Sehr häufige Nebenwirkungen (können bei mehr als 1 von 10 Patienten auftreten):
• Infusionsreaktionen, die zum Zeitpunkt der Infusion oder innerhalb von 24 Stunden nach der Infusion auftreten können: Kopfschmerz, Ausschlag, Fieber, Unwohlsein, Quaddeln, Jucken, Rötung des Gesichts und des Halses, Müdigkeit
• Infektionen: Atemwegsinfektionen, wie etwa Erkältungen oder Nasennebenhöhlenentzündungen,
Blasenentzündung
• Reduzierte Anzahl der weißen Blutkörperchen (Lymphozyten)
Häufige Nebenwirkungen (können bei bis zu 1 von 10 Patienten auftreten):
• Infusionsreaktionen, die zum Zeitpunkt der Infusion oder innerhalb von 24 Stunden nach der Infusion auftreten können: Veränderung der Herzfrequenz, Verdauungsstörungen, Schüttelfrost, Brustbeschwerden, Schmerz, Schwindelgefühl, Geschmacksstörungen, Schlafstörungen, Schwierigkeiten beim Atmen oder Kurzatmigkeit, Ausschlag am Körper, niedriger Blutdruck
• Infektionen: Husten, Ohreninfektion, grippeartige Erkrankung, Bronchitis, Lungenentzündung, Pilzinfektionen in Mund oder Scheide, Gürtelrose, Windpocken, oraler Herpes, geschwollene oder vergrößerte Lymphknoten
• Schmerzen an der Infusionsstelle, Schmerzen im Rücken, Nacken oder in den Armen oder Beinen, Muskelschmerz, Muskelkrämpfe, Gelenkschmerz, Mund- oder Halsschmerzen
• Entzündung von Mund/Zahnfleisch/Zunge
• Allgemeines Unwohlsein/Krankheitsgefühl, Schwäche, Erbrechen, Durchfall, Unterleibsschmerz, Magen-Darm-Grippe
• Sodbrennen
• Veränderungen, die bei Untersuchungen festgestellt werden können: Blut oder Eiweiß im Urin, erniedrigte Herzfrequenz, unregelmäßiger oder anomaler Herzschlag, erhöhter Blutdruck
• MS-Schub
• Zittern, Taubheitsgefühl, Brennen oder Kribbeln
• Schilddrüsenüber- oder Schilddrüsenunterfunktion oder Struma (Anschwellen der Schilddrüse im
Hals)
• Anschwellen von Armen und/oder Beinen
• Sehstörungen
• Angstgefühl
• Stärkere oder länger andauernde Menstruation als normal oder unregelmäßige Menstruation
• Akne, Hautrötung, übermäßiges Schwitzen
• Nasenbluten, blaue Flecken
• Haarausfall
Gelegentliche Nebenwirkungen (können bei bis zu 1 von 100 Patienten auftreten)
• Infektionen: Genitaler Herpes, Augeninfektion, Zahninfektion
• Probleme mit der Blutgerinnung, Anämie
• Fußpilz
• Veränderter Vaginalabstrich
• Depression
• Überempfindlichkeit für Berührungsreize
• Schluckbeschwerden
• Schluckauf
• Verringertes Gewicht
• Verstopfung
• Zahnfleischbluten
• Abnormale Leberwerte
• Blasen
Legen Sie die Patientenkarte und diese Packungsbeilage jedem Arzt vor, der Sie behandelt, nicht nur Ihrem Neurologen.
Sie finden diese Informationen auch in der Patientenkarte und im Leitfaden für Patienten, die Ihnen von Ihrem Arzt ausgehändigt wurden.
Meldung von Nebenwirkungen
Wenn Sie Nebenwirkungen bemerken, wenden Sie sich an Ihren Arzt. Dies gilt auch für Nebenwirkungen, die nicht in dieser Packungsbeilage angegeben sind. Sie können Nebenwirkungen auch direkt über das in Anhang V aufgeführte nationale Meldesystem anzeigen. Indem Sie Nebenwirkungen melden, können Sie dazu beitragen, dass mehr Informationen über die Sicherheit dieses Arzneimittels zur Verfügung gestellt werden.
5. Wie ist LEMTRADA aufzubewahren?
Bewahren Sie dieses Arzneimittel für Kinder unzugänglich auf.
Sie dürfen dieses Arzneimittel nach dem auf dem Umkarton und dem Etikett nach „Verwendbar bis“ oder „EXP“ angegebenen Verfalldatum nicht mehr verwenden. Das Verfallsdatum bezieht sich auf den letzten Tag des angegebenen Monats.
Im Kühlschrank lagern (2°C - 8°C).
Nicht einfrieren.
In der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen.
Es wird aufgrund eines möglichen Risikos einer mikrobiellen Kontamination empfohlen, das Arzneimittel unmittelbar nach der Verdünnung zu verwenden. Bei einer nicht sofortigen Anwendung ist der Anwender für die Einhaltung einer entsprechenden Lagerungsdauer und entsprechender Lagerbedingungen unter Schutz vor Lichteinwirkung verantwortlich (nicht länger als 8 Stunden bei 2°C bis 8°C).
Sie dürfen dieses Arzneimittel nicht verwenden, wenn Sie Teilchen in der Flüssigkeit und/oder eine Verfärbung der Flüssigkeit in der Durchstechflasche bemerken.
Entsorgen Sie Arzneimittel nicht im Abwasser. Sie tragen damit zum Schutz der Umwelt bei.
6. Inhalt der Packung und weitere Informationen
Was LEMTRADA enthält Der Wirkstoff ist Alemtuzumab.
Jede Durchstechflasche enthält 12 mg Alemtuzumab in 1,2 ml.
Die sonstigen Bestandteile sind:
• Dinatriumphosphat-Dihydrat (E339)
• Dinatriumedetat-Dihydrat
• Kaliumchlorid (E508)
• Kaliumdihydrogenphosphat (E340)
• Polysorbat 80 (E433)
• Natriumchlorid
• Wasser für Injektionszwecke
Wie LEMTRADA aussieht und Inhalt der Packung
LEMTRADA ist ein klares, farbloses bis leicht gelbes Konzentrat zur Herstellung einer Infusionslösung (steriles Konzentrat) in einer Durchstechflasche aus Glas mit Stopfen.
In jedem Umkarton befindet sich 1 Flasche.
Pharmazeutischer Unternehmer
Genzyme Therapeutics Ltd, 4620 Kingsgate, Cascade Way, Oxford Business Park South, Oxford, OX4 2SU, Vereinigtes Königreich.
Hersteller
Genzyme Ltd., 37 Hollands Road, Haverhill, Suffolk CB9 8PU, Vereinigtes Königreich.
Genzyme Ireland Limited, IDA Industrial Park, Old Kilmeaden Road, Waterford, Irland.
Falls Sie weitere Informationen über das Arzneimittel wünschen, setzen Sie sich bitte mit dem örtlichen Vertreter des Pharmazeutischen Unternehmers in Verbindung.
|
Belgie/Belgique/Belgien/ Luxemburg/Luxembourg Sanofi Belgium Tel/Tel: + 32 2 710 54 00 |
Lietuva UAB „SANOFI-AVENTIS LIETUVA“ Tel. +370 5 275 5224 |
|
Btnrapna Sanofi-Aventis Bulgaria EOOD Ten: +359 2 9705300 |
Magyarorszag sanofi-aventis Zrt Tel: +36 1 505 0050 |
|
Ceska republika sanofi-aventis, s.r.o. Tel: +420 233086 111 |
Malta Sanofi-Aventis Malta Ltd Tel: +356 21493022 |
|
Danmark sanofi-aventis Denmark A/S Tlf: +45 45 16 70 00 |
Nederland Genzyme Europe B.V. Tel: +31 35 699 1200 |
|
Deutschland Genzyme Therapeutics Ltd. Tel: +49 (0) 6102 3674 451 |
Norge sanofi-aventis Norge AS Tlf: + 47 67 10 71 00 |
|
Eesti sanofi-aventis Estonia OÜ Tel. +372 6 273 488 |
Österreich sanofi-aventis GmbH Tel: + 43 1 80 185 - 0 |
|
Ekkdöa/Kunpo^ sanofi-aventis AEBE (ELLaSa) T pL: +30 210 900 1600 |
Polska sanofi-aventis Sp. z o.o. Tel.: +48 22 280 00 00 |
|
Espana Genzyme, S.L.U. Tel: +34 93 485 94 00 sanofi-aventis, S.A. Tel: +34 93 485 94 00 |
Portugal Sanofi - Produtos Farmaceuticos, Lda. Tel: +351 21 422 0100 |
|
France Genzyme S.A.S. Tel : +33 (0) 825 825 863 |
Romania sanofi-aventis Romania S.R.L. Tel: +40 (0) 21 317 31 36 |
Latvija
sanofi-aventis Latvia SIA Tel: +371 67 33 24 51
Hrvatska
sanofi-aventis Croatia d.o.o.
Tel: +385 1 6003 400
Island
Vistor hf.
Simi: +354 535 7000 Ireland
Genzyme Therapeutics Ltd. (United Kingdom) Tel: +44 (0) 1865 405200
Slovenija
sanofi-aventis d.o.o.
Tel: +386 1 560 4800
Slovenska republika
sanofi-aventis Pharma Slovakia s.r.o. Tel.: +421 2 33 100 100
Suomi/Finland
sanofi-aventis Oy Puh/Tel: + 358 201 200 300
Italia
Genzyme Srl Tel: +39 059 349 811
Sverige
sanofi-aventis AB
Tel: +46 (0)8 634 50 00
United Kingdom
Genzyme Therapeutics Ltd. (United Kingdom)
Tel: +44 (0) 1865 405200
Diese Packungsbeilage wurde zuletzt überarbeitet im Weitere Informationsquellen
Zur Unterstützung von Patientenschulungen zu potentiellen Nebenwirkungen und Anweisungen, was im Falle bestimmter Nebenwirkungen zu tun ist, sind die folgenden Materialien zur Risikominimierung verfügbar:
1. Patientenkarte: Für den Patienten zur Vorlage bei anderem medizinischen Fachpersonal, um auf die
Anwendung von LEMTRADA bei diesem Patienten hinzuweisen
2. Leitfaden für Patienten: Für weitere Informationen zu Autoimmunreaktionen, Infektionen u. a.
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur http://www.ema.europa.eu/ verfügbar.
Die folgenden Informationen sind nur für medizinisches Fachpersonal bestimmt:
Informationen zur Risikominimierung - Autoimmunerkrankungen
• Es ist äußerst wichtig, dass Ihr Patient die Verpflichtung zur Durchführung regelmäßiger Untersuchungen (über 4 Jahre nach der letzten Infusion) versteht, auch wenn er keine Symptome aufweist und die MS gut kontrolliert ist.
• Sie müssen zusammen mit Ihrem Patienten die regelmäßigen Kontrolluntersuchungen planen und organisieren.
• Hält der Patient die Nachbeobachtungstermine nicht ein, muss er eventuell erneut auf die Risiken von Versäumnissen bei den geplanten Kontrolluntersuchungen hingewiesen werden.
• Sie sollten die Untersuchungsergebnisse überprüfen und auf Symptome von Nebenwirkungen achten.
• Gehen Sie den LEMTRADA-Leitfaden für Patienten und diese Packungsbeilage mit Ihrem Patienten durch. Erinnern Sie den Patienten daran, auf Symptome im Zusammenhang mit bestehenden Autoimmunerkrankungen zu achten und ärztliche Hilfe aufzusuchen, wenn Fragen bestehen.
Schulungsmaterialien für medizinisches Fachpersonal sind ebenfalls erhältlich:
• LEMTRADA-Leitfaden für Ärzte
• LEMTRADA-Schulungsmodul
• LEMTRADA-Checkliste für verordnende Ärzte
Lesen Sie die Fachinformation (Zusammenfassung der Merkmale des Arzneimittels, verfügbar auf der o. g.
Website der EMA) für weitere Informationen.
Informationen zur Vorbereitung der Verabreichung von LEMTRADA und zur Überwachung des Patienten
• Die Patienten sollten in den ersten 3 Tagen einer jeden Behandlungsphase unmittelbar vor der LEMTRADA-Infusion mit Kortikosteroiden vorbehandelt werden. Es kann auch eine Vorbehandlung mit Antihistaminika und/oder Antipyretika vor der Verabreichung von LEMTRADA in Erwägung gezogen werden.
• Allen Patienten sollte während und 1 Monat lang nach der Behandlung ein orales Arzneimittel gegen Herpes verabreicht werden. In klinischen Studien wurde den Patienten zweimal täglich Aciclovir 200 mg oder ein äquivalentes Arzneimittel verabreicht.
• Führen Sie die Eingangsuntersuchungen und Screenings wie unter Abschnitt 4 der Zusammenfassung der Merkmale des Arzneimittels (Fachinformation) beschrieben durch.
• Der Inhalt der Durchstechflasche sollte vor der Anwendung auf Partikel und Verfärbungen überprüft werden. Nicht verwenden, wenn Partikel vorhanden sind oder sich das Konzentrat verfärbt hat.
DIE DURCHSTECHFLASCHEN VOR DER ANWENDUNG NICHT SCHÜTTELN.
• Nehmen Sie unter Anwendung aseptischer Methoden 1,2 ml LEMTRADA aus der Durchstechflasche in eine Spritze auf und geben Sie es in 100 ml 0,9%ige NatriumchloridInfusionslösung (9 mg/ml) oder in 5%ige Glukose-Infusionslösung. Der Beutel sollte vorsichtig geschwenkt werden, um die Lösung zu mischen. Es ist Vorsicht geboten, um die Sterilität der zubereiteten Lösung sicherzustellen, insbesondere weil diese keine Konservierungsmittel enthält.
• Verabreichen Sie die LEMTRADA-Infusionslösung intravenös über einen Zeitraum von etwa 4 Stunden.
• Es sollten keine anderen Arzneimittel zur LEMTRADA-Infusionslösung hinzugegeben oder gleichzeitig durch denselben intravenösen Zugang verabreicht werden.
• Es wird aufgrund eines möglichen Risikos einer mikrobiellen Kontamination empfohlen, das Arzneimittel unmittelbar nach der Verdünnung zu verwenden. Bei einer nicht sofortigen Anwendung ist der Anwender für die Einhaltung einer entsprechenden Lagerungsdauer und entsprechender Lagerbedingungen unter Schutz vor Lichteinwirkung verantwortlich (nicht länger als 8 Stunden bei 2°C bis 8°C).
• Die Verfahren zur korrekten Handhabung und Beseitigung sollten beachtet werden. Verschüttetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu entsorgen.
• Nach jeder Infusion sollte der Patient 2 Stunden lang in Hinblick auf infusionsbedingte Reaktionen überwacht werden. Bei Bedarf kann eine symptomatische Behandlung eingeleitet werden - siehe
Zusammenfassung der Merkmale des Arzneimittels (Fachinformation). Führen Sie weiterhin jeden Monat bis 4 Jahre nach der letzten Infusion Untersuchungen des Patienten in Hinblick auf Autoimmunerkrankungen durch. Für weitere Informationen lesen Sie den LEMTRADA-Leitfaden für Ärzte oder lesen Sie die Fachinformation (Zusammenfassung der Merkmale des Arzneimittels), die auf der o. g. Website der EMA verfügbar ist.
ANHANGIV
WISSENSCHAFTLICHE SCHLUSSFOLGERUNGEN UND GRÜNDE FÜR DIE ÄNDERUNG DER BEDINGUNGEN DER GENEHMIGUNG(EN) FÜR DAS INVERKEHRBRINGEN
Wissenschaftliche Schlussfolgerungen
Der CHMP ist unter Berücksichtigung des PRAC-Beurteilungsberichts zum PSUR für Alemtuzumab zu den folgenden wissenschaftlichen Schlussfolgerungen gelangt:
Listeriose/Listerienmeningitis
Arzneimittel mit Immunsystem-modulierenden Eigenschaften, wie z.B. Lemtrada, können mit einem erhöhten Risiko für opportunistische Infektionen assoziiert sein. Insgesamt wurden 5 Fälle aus ganz Europa identifiziert. Ein MS-Patient, der im Rahmen der klinischen Studie CAMMS223 mit Alemtuzumab behandelt wurde, entwickelte eine Listerienmeningistis. 4 Fälle von systemischer Listeriose oder Meningitis, verursacht durch Listeria monocytogenes, wurden nach Markteinführung berichtet.
Bradykardie als infusionsassoziierte Nebenwirkung
In klinischen Studien wurden 71 Fälle von Bradykardie (in 55 Patienten) gemeldet, von denen zwei als schwerwiegend und die restlichen als nicht schwerwiegend eingestuft wurden. Insgesamt nahmen 1.505 Alemtuzumab-Patienten an diesen Studien teil. Zusätzlich wurden 39 Fälle von Bradykardie, von denen 8 als schwerwiegend und die restlichen als nicht schwerwiegend eingestuft wurden, nach der Markteinführung von Alemtuzumab ab dem 01. Mai 2015 berichtet. Jede der 10 schwerwiegenden Bradykardien trat als infusionsassoziierte Reaktion auf.
Nach Bewertung der im PSUR vorliegenden Daten hält das PRAC eine Änderung der Produktinformationen für das Alemtuzumab-enthaltende Arzneimittel für berechtigt. Der Abschnitt 4.4 der Zusammenfassung der Merkmale des Arzneimittels (Fachinformation) und die betroffenen Abschnitte der Packungsbeilage wurden überarbeitet.
Der CHMP stimmt den wissenschaftlichen Schlussfolgerungen des PRAC zu.
Gründe für die Änderung der Bedingungen der Genehmigung(en) für das Inverkehrbringen
Der CHMP ist auf der Grundlage der wissenschaftlichen Schlussfolgerungen für Alemtuzumab der Auffassung, dass das Nutzen-Risiko-Verhältnis des Arzneimittels, vorbehaltlich der vorgeschlagenen Änderungen der Produktinformation, unverändert ist.
Der CHMP empfiehlt, die Bedingungen der Genehmigung für das Inverkehrbringen zu ändern.
45