Anbinex
ZUSAMMENFASSUNG DER MERKMALE DES ARZNEIMITTELS
1. BEZEICHNUNG DES ARZNEIMITTELS
Anbinex® 500 I.E. und Anbinex® 1000 I.E., Trockensubstanz und Lösungsmittel zur Herstellung einer Injektionslösung Wirkstoff: Antithrombin vom Menschen
2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
Anbinex® ist eine lyophilisierte Trockensubstanz zur Herstellung einer Injektionslösung, die 500 I.E. bzw. 1000 I.E. Antithrombin vom Menschen pro Durchstechflasche enthält.
Anbinex® 500 I.E. enthält nach Rekonstitution im beigepackten Lösungsmittel
(10 ml Wasser für Injektionszwecke) ungefähr 50 I.E. Antithrombin vom Menschen pro ml.
Anbinex® 1000 I.E. enthält nach Rekonstitution im beigepackten Lösungsmittel
(20 ml Wasser für Injektionszwecke) ungefähr 50 I.E. Antithrombin vom Menschen pro ml.
Die Bestimmung der Aktivität (I.E.) wird mittels der chromogenen Methode gemäß Europäischem Arzneibuch durchgeführt. Die spezifische Aktivität von Anbinex® beträgt mindestens 5 I.E./mg Protein.
Sonstige(r) Bestandteil(e) mit bekannter Wirkung:
Anbinex® 500 I.E. enthält 1,45 mmol Natrium pro 10 ml Lösung.
Anbinex® 1000 I.E. enthält 2,90 mmol Natrium pro 20 ml Lösung.
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
3. DARREICHUNGSFORM
Trockensubstanz und Lösungsmittel zur Herstellung einer Injektionslösung
Flasche mit weißer, hygroskopischer, bröckeliger Trockensubstanz und vorgefüllte Spritze mit Wasser für Injektionszwecke.
4. KLINISCHE ANGABEN
4.1 Anwendungsgebiete
- Angeborener Mangel an Antithrombin:
a. Prophylaxe von tiefer Venenthrombose und Thromboembolie in klinischen Risikosituationen (insbesondere während operativer Eingriffe oder während Schwangerschaft und Geburt) in Verbindung mit Heparin-Gabe soweit dies indiziert ist.
b. Verhinderung des Fortschreitens von tiefer Venenthrombose und Thromboembolie in Verbindung mit Heparin-Gabe soweit dies indiziert ist.
- Erworbener Mangel an Antithrombin
4.2 Dosierung und Art der Anwendung
Die Behandlung sollte unter Überwachung eines Arztes erfolgen, der Erfahrung in der Therapie von Patienten mit Antithrombin-Mangel besitzt.
Dosierung
Bei angeborenem Mangel sollte die Dosierung unter Berücksichtigung der Familienanamnese in Bezug auf thromboembolische Ereignisse, der tatsächlichen klinischen Risikofaktoren und der Laboruntersuchungen individuell für den Patienten angepasst werden.
Die Dosierung und Dauer der Substitutionstherapie bei erworbenem Mangel hängt vom Antithrombin-Plasmaspiegel, von Anzeichen erhöhten Verbrauchs, der zu Grunde liegenden Erkrankung und der Schwere der klinischen Symptome ab. Die Menge und Häufigkeit der Verabreichungen sollte sich im Einzelfall stets nach der klinischen Wirksamkeit und den Laborergebnissen richten.
Die Menge der verabreichten Antithrombin-Einheiten wird in Internationalen Einheiten (I.E.) angegeben, die vom aktuellen WHO-Standard für Antithrombin-Produkte abgeleitet sind. Die Antithrombin-Aktivität im Plasma wird entweder als Prozentsatz (relativ zu normalem menschlichem Plasma) oder in Internationalen Einheiten (relativ zum Internationalen Standard für Antithrombin im Plasma) angegeben.
Eine Internationale Einheit (I.E.) Antithrombin entspricht der Antithrombin-Aktivität in einem Milliliter normalen menschlichen Plasmas. Die Berechnung der benötigten AntithrombinDosierung basiert auf der empirischen Beobachtung, dass eine Internationale Einheit (I.E.) Antithrombin pro kg Körpergewicht die Antithrombin-Aktivität im Plasma um etwa 1,1 bis 1,6 % erhöht.
Die Anfangsdosis wird nach folgender Formel berechnet:
Erforderliche Einheiten = Körpergewicht (kg) x ( 100 - aktuelle Antithrombin-Aktivität [%]) x 0,8
Die angestrebte anfängliche Antithrombin-Aktivität hängt vom klinischen Zustand des Patienten ab. Ist die Indikation für die Antithrombin-Substitution eindeutig geklärt, sollte die Dosierung ausreichend sein um die angestrebte Antithrombin-Aktivität zu erreichen und einen wirksamen Spiegel zu erhalten. Die Dosierung sollte auf Basis der Bestimmung der Antithrombin-Aktivität, welche bis zur Stabilisierung des Patienten mindestens zweimal täglich, danach einmal täglich (bevorzugt unmittelbar vor der nächsten Injektion) durchgeführt werden soll, festgesetzt und überwacht werden. Bei einer Änderung der Dosierung sollten sowohl Anzeichen eines erhöhten Antithrombin-Verbrauchs als auch der klinische Verlauf berücksichtigt werden. Für die Dauer der Behandlung sollte eine Antithrombin-Aktivität über 80 % erreicht werden, es sei denn klinische Besonderheiten sprechen für einen anderen Wirkspiegel.
Die übliche Anfangsdosierung bei angeborenem Mangel liegt bei 30 bis 50 I.E./kg.
Danach sollte die Dosis und Häufigkeit sowie die Dauer der Behandlung an die Laborwerte und die klinische Situation angepasst werden.
Kinder und Jugendliche
Es liegen nicht genügend Daten aus klinischen Studien über die Behandlung von Kindern unter 6 Jahren vor.
Art der Anwendung
Das Auflösen der Zubereitung erfolgt wie in Abschnitt 6.6. beschrieben.
Anbinex® soll intravenös mit einer maximalen Infusionsgeschwindigkeit von 0,08 ml/kg/min verabreicht werden.
4.3 Gegenanzeigen
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Aufgrund von Erfahrungen aus klinischen Prüfungen kann die Anwendung von Antithrombin zur Behandlung von IRDS (Infant Respiratory Distress Syndrome) bei Frühgeborenen nicht empfohlen werden.
Wie bei jedem intravenös zu verabreichenden proteinhaltigen Produkt sind allergische Überempfindlichkeitsreaktionen möglich.
Die Patienten müssen engmaschig überwacht werden und sorgfältig auf Symptome während der Infusion beobachtet werden.
Die Patienten sollten über frühe Zeichen von Überempfindlichkeitsreaktionen, wie Ausschlag, generalisierte Urtikaria, Engegefühl in der Brust, Giemen, Hypotonie und Anaphylaxie informiert werden. Wenn diese Symptome auftreten, sollen sie sofort ihren Arzt informieren.
Beim Auftreten von Schocksymptomen, sind die aktuellen medizinischen Empfehlungen für Schocktherapie zu befolgen.
Standardmaßnahmen zur Verhütung von Infektionen durch Arzneimittel aus menschlichem Blut oder Plasma beinhalten die Auswahl der Spender, die Untersuchung der einzelnen Spenden und der Plasmapools auf spezifische Infektionsmarker und die Aufnahme von Herstellungsschritten zur wirksamen Inaktivierung/Eliminierung von Viren. Trotz dieser Maßnahmen kann bei der Anwendung von Arzneimitteln, die aus menschlichem Blut oder Plasma hergestellt werden, die Möglichkeit der Übertragung einer Infektion nicht vollständig ausgeschlossen werden. Dies trifft auch für alle unbekannten oder neu auftauchenden Viren oder anderen Krankheitserreger zu.
Die durchgeführten Maßnahmen (Pasteurisierung, Nanofiltration) werden bei umhüllten Viren wie dem Human Immunodeficiency Virus (HIV), dem Hepatitis-B-Virus (HBV) und dem Hepatitis-C-Virus (HCV) sowie bei dem nicht-umhüllten Hepatitis-A-Virus als wirksam erachtet. Die Maßnahmen können von begrenzter Wirksamkeit gegen nicht-umhüllte Viren wie Parvovirus B19 sein.
Eine Parvovirus-B19-Infektion während der Schwangerschaft kann schwerwiegende Folgen haben (fetale Infektion). Ebenfalls gefährdet sind Personen mit Immunschwäche oder gesteigerter Erythropoese (z.B. bei hämolytischer Anämie).
Bei Patienten, die regelmäßig/wiederholt Antithrombin-Produkte aus menschlichem Plasma erhalten, sollte eine entsprechende Impfung (Hepatitis A und Hepatitis B) in Erwägung gezogen werden.
Es wird darauf hingewiesen, dass gemäß Transfusionsgesetz bei jeder Behandlung eines Patienten mit Anbinex® der Produktname und die Chargenbezeichnung dokumentiert werden müssen, um eine Verbindung zwischen dem Patienten und der verwendeten Charge sicherzustellen.
Folgende Laboruntersuchungen sind bei gemeinsamer Gabe von Antithrombin und Heparin sinnvoll:
- Um die Heparindosis anzupassen und zur Vermeidung einer überschießenden Gerinnungshemmung, sollten regelmäßige Kontrollen (APPT und - wo sinnvoll - anti-FXa-Aktivität) in engen Intervallen und insbesondere während der ersten Minuten/Stunden nach Gabe von Antithrombin durchgeführt werden.
- Tägliche Bestimmung des Antithrombin-Spiegels zur Anpassung der individuellen Dosis, da eine längere Behandlung mit nicht-fraktioniertem Heparin zu einer Verringerung des Antithrombin-Spiegels führen kann.
Anbinex® 500 I.E. enthält 1,45 mmol Natrium pro 10 ml Lösung. Anbinex® 1000 I.E. enthält 2,90 mmol Natrium pro 20 ml Lösung. Dies muss bei Patienten, die eine natriumarme Diät einhalten müssen, berücksichtigt werden.
4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Heparin: Antithrombin-Substitution während der Behandlung mit therapeutischen Dosen von Heparin erhöht das Risiko von Blutungen. Die Wirkung von Antithrombin wird durch Heparin deutlich verstärkt. Die Halbwertszeit von Antithrombin kann bei gleichzeitiger Behandlung mit Heparin aufgrund des erhöhten Antithrombin-Verbrauchs stark erniedrigt sein. Die kombinierte Gabe von Heparin und Antithrombin muss bei Patienten mit erhöhtem Risiko von Blutungen deshalb engmaschig überprüft werden.
4.6 Fertilität, Schwangerschaft und Stillzeit
Die Erfahrung zur Sicherheit des Einsatzes von Antithrombin-Produkten während der Schwangerschaft ist begrenzt.
Unter Berücksichtigung des erhöhten Risikos thromboembolischer Ereignisse während der Schwangerschaft kann Anbinex® schwangeren und stillenden Frauen mit Antithrombin-Mangel bei eindeutiger Indikationsstellung verabreicht werden.
4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Anbinex® hat keinen Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
4.8 Nebenwirkungen
In klinischen Studien mit Anbinex® bzw. dem Vorläuferprodukt wurden keine Nebenwirkungen berichtet.
Generell können bei der Behandlung mit Antithrombin vom Menschen folgende Nebenwirkungen auftreten: Überempfindlichkeit oder allergische Reaktionen (wie z.B. Quincke-Ödem, Brennen und Stechen an der Einstichstelle, Schüttelfrost, Hitzegefühl, generalisierte Urtikaria, Kopfschmerz, Quaddeln, Hypotonie, Lethargie, Übelkeit,
Ruhelosigkeit, Tachykardie, Engegefühl in der Brust, Kribbeln, Erbrechen, Giemen) wurden selten beobachtet. Diese Reaktionen können in einigen Fällen zu schwerer Anaphylaxie (mit Schock) führen. Beim Auftreten von Schocksymptomen, sind die aktuellen medizinischen Empfehlungen für Schocktherapie zu befolgen.
In seltenen Fällen wurde Fieber beobachtet.
Informationen zur Sicherheit hinsichtlich übertragbarer Krankheiten: siehe 4.4.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel, Paul-Ehrlich-Institut, Paul-Ehrlich-Straße 51-59, 63225 Langen, Tel: +49 6103 77 0, Fax: +49 6103 77 1234, Website: www.pei.de anzuzeigen.
4.9 Überdosierung
Es wurden keine Fälle von Überdosierung berichtet.
5.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Antithrombotika: Heparin-Gruppe ATC-Code: B01AB02
Antithrombin ist ein Glykoprotein von 58 kD und 432 Aminosäuren und gehört zur Gruppe der Serpine (Serinprotease-Inhibitoren). Es ist einer der wichtigsten Inhibitoren der Blutgerinnung. Die am stärksten inhibierten Faktoren sind Thrombin und Faktor Xa, aber auch Faktoren der Kontaktaktivierung, des intrinsischen Systems sowie der Faktor VIIa/Gewebefaktor-Komplex werden gehemmt. Die Antithrombin-Aktivität wird durch Heparin deutlich erhöht und die antikoagulatorische Wirkung von Heparin ist abhängig von der Gegenwart Antithrombins.
Antithrombin enthält zwei funktionell wichtige Domänen. Die erste enthält das reaktive Zentrum und dient als Spaltstelle für Proteinasen wie Thrombin, eine Voraussetzung zur Bildung eines stabilen Proteinase-Inhibitor-Komplexes. Die zweite ist ein Glykosaminoglykan-Bindungsbereich, der für die Wechselwirkung mit Heparin und verwandten Substanzen, welche die Hemmung von Thrombin verstärken, zuständig ist. Die Komplexe aus Inhibitor und Gerinnungsenzym werden über das retikuloendotheliale System abgebaut.
Die Antithrombin-Aktivität bei Erwachsenen beträgt 80-120 % und bei Neugeborenen ca. 40-60 %.
5.2 Pharmakokinetische Eigenschaften
Pharmakokinetische Studien zu Antithrombin zeigen eine mittlere Halbwertszeit von etwa 3 Tagen. Die Halbwertszeit kann bei gleichzeitiger Heparinbehandlung auf ca. 1,5 Tage absinken. In Zuständen hohen Verbrauchs kann sie sogar auf Stunden abfallen.
Folgende Ergebnisse wurden in einer klinischen Studie mit Anbinex® bei Patienten mit angeborenem Antithrombin-Mangel anhand eines unabhängigen Analysenmodells ermittelt:
- Incremental recovery: 1,3 ± 0,2 I.E./dl/I.E./kg mit einem Bereich von 1,1 bis 1,6 % (basierend auf der Aktivitätsbestimmung)
- Area under curve (AUC): 66,5 ± 15,4 I.E. h/l
- Terminale Halbwertszeit: 98,1 ± 45,0 h (basierend auf der Aktivitätsbestimmung)
- Mean residence time (MRT): 121,7 ± 52,1 h
- Clearance: 0,931 ± 0,214 ml/h/kg.
5.3 Präklinische Daten zur Sicherheit
Antithrombin ist ein normaler Bestandteil des menschlichen Plasmas.
Akute Toxizitätsstudien sind von geringer Aussagekraft und erlauben keine Abschätzung der toxischen oder tödlichen Dosis oder der Dosis-Wirkungs-Beziehung.
Studien zur chronischen Toxizität bei Tieren sind wegen der Bildung von Antikörpern nicht praktikabel.
Es wurden keine Anzeichen akuter Toxizität in Tiermodellen beschrieben.
6. PHARMAZEUTISCHE ANGABEN 6.1 Liste der sonstigen Bestandteile
Natriumchlorid, Natriumcitrat, D-Mannitol, Wasser für Injektionszwecke (Lösungsmittel).
6.2 Inkompatibilitäten
Da keine Kompatibilitätsstudien durchgeführt wurden, darf diese Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
6.3 Dauer der Haltbarkeit
Trockensubstanz:
3 Jahre.
Rekonstituierte Lösung:
Die chemische und physikalische Gebrauchsstabilität wurde für 12 Stunden bei 25°C nachgewiesen.
Aus mikrobiologischer Sicht sollte das Produkt sofort verwendet werden. Wird das aufgelöste Produkt nicht sofort verwendet, so liegt die Verantwortung für die Gebrauchsdauer und die Bedingungen vor der Anwendung beim Anwender.
6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Nicht über 30 °C lagern. Nicht einfrieren.
Aufbewahrungsbedingungen nach Rekonstitution des Arzneimittels, siehe Abschnitt 6.3.
6.5 Art und Inhalt des Behältnisses Anbinex® 500 I.E.:
Durchstechflasche (Typ II Glas, Brombutylstopfen) mit 500 I.E. Antithrombin (Trockensubstanz) und Glasspritze (Typ I Glas, Brombutylstopfen) mit 10 ml Wasser für Injektionszwecke (Lösungsmittel).
Anbinex® 1000 I.E.:
Durchstechflasche (Typ II Glas, Brombutylstopfen) mit 1000 I.E. Antithrombin (Trockensubstanz) und Glasspritze (Typ I Glas, Brombutylstopfen) mit 20 ml Wasser für Injektionszwecke (Lösungsmittel).
Beigefügte Medizinprodukte zur Rekonstitution von Anbinex® sind:
Microfilter, Transferadapter.
Jede Einzelpackung enthält: 1 Flasche mit Lyophilisat, 1 vorgefüllte Spritze mit Wasser für Injektionszwecke und Zubehör zur Rekonstitution.
6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung
Nach Ablauf des auf dem Etikett angegebenen Verfalldatums darf Anbinex® nicht mehr verwendet werden.
Das rekonstituierte Produkt sollte visuell auf Partikel und Verfärbung geprüft werden. Die Lösung sollte klar oder leicht opaleszent sein. Keine Lösungen verwenden, die trübe sind oder einen Bodensatz aufweisen.
Nicht verwendete Lösung darf nicht zur späteren Anwendung aufbewahrt werden.
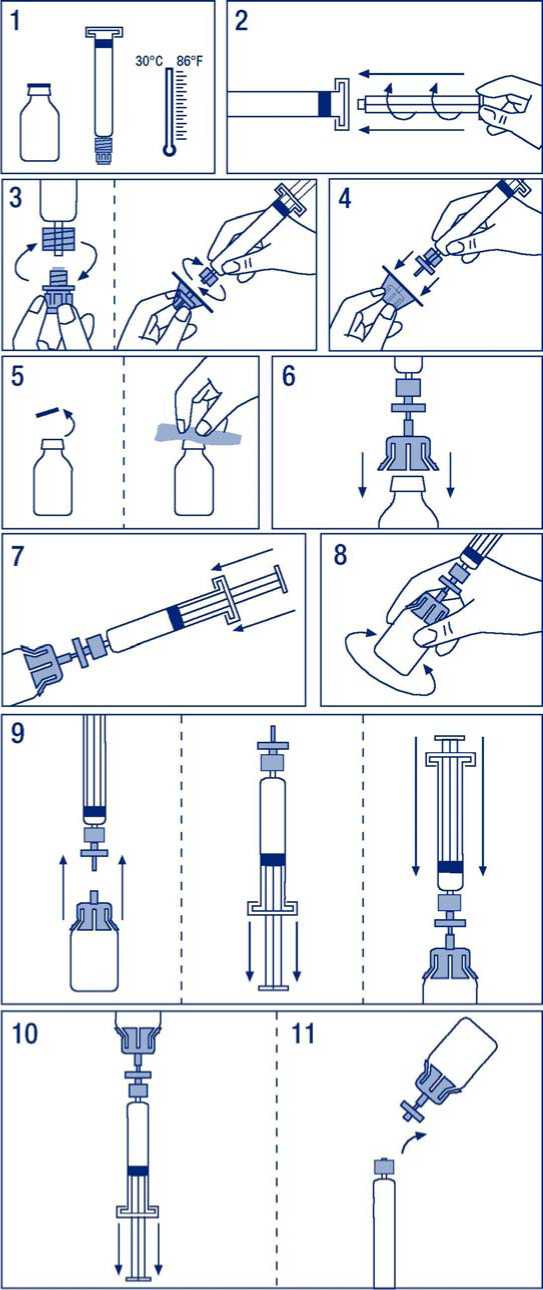
Herstellung der Injektionslösung:
1. Produktflasche und Spritze auf Raumtemperatur bringen.
2. Kolben in die Spritze mit dem Lösungsmittel eindrehen, wobei die Plastikmanschette in diesem Bereich unter keinen Umständen entfernt werden darf.
3. Folie von der Filterverpackung abziehen. Verschlusskappe von der Spritzenspitze entfernen und Spritze auf den Filter drehen.
4. Transferadapter aus der Verpackung nehmen und auf die Spritze mit dem Filter setzen.
5. Kunststoffdeckel von der Produktflasche entfernen und den Gummistopfen desinfizieren.
6. Gummistopfen der Produktflasche mit der Kanüle des Transferadapters durchstechen.
7. Das gesamte Lösungsmittel wird nun aus der Spritze in die Produktflasche überführt.
8. Spritze mit Produktflasche vorsichtig schwenken bis das Lyophilisat vollständig gelöst ist.
9. Spritze mit Filter von der Produktflasche mit Transferadapter trennen. Den Kolben der Spritze zurückziehen, um eine dem Gesamtvolumen der Lösung vergleichbare Menge Luft aufzuziehen. Danach wieder Spritze mit Filter und Produktflasche mit Transferadapter verbinden und Luft injizieren.
10. Produktflasche mit aufgesetzter Spritze umdrehen und Lösung in die Spritze aufziehen.
11. Spritze (ohne Filter!) abnehmen und Lösung langsam intravenös injizieren. Die Infusionsgeschwindigkeit sollte 0,08 ml/kg/min nicht überschreiten.
Sämtliche Teile dürfen nicht wieder verwendet werden.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
INHABER DER ZULASSUNG UND HERSTELLER
7.
Zulassungsinhaber:
Grifols Deutschland GmbH Lyoner Straße 15 60528 Frankfurt Tel.: 069/660 593 100 Fax: 069/660 593 110
Hersteller:
Instituto Grifols, S.A.
Can Guasc, 2 - Parets del Valles E-08150 Barcelona
8. ZULASSUNGSNUMMER
7812.00.00
9. DATUM DER VERLÄNGERUNG DER ZULASSUNG
Datum der letzten Verlängerung der Zulassung:13.Juni 2006
10. STAND DER INFORMATION
07.2014
11. VERSCHREIBUNGSSTATUS/APOTHEKENPFLICHT Verschreibungspflichtig
12. HERKUNFTSLAND DES BLUTPLASMAS
Das zur Herstellung von Anbinex® verwendete Blutplasma stammt aus den USA, Tschechien oder der Slowakei.
Seite 9 von 9