Ben-U-Ron 500Mg
Zusammenfassung der Merkmale des Arzneimittels
Fachinformation
1. Bezeichnung des Arzneimittels ben-u-ron® 500 mg Zäpfchen
2. Qualitative und quantitative Zusammensetzung
Wirkstoff: 1 Zäpfchen enthält 500 mg Paracetamol.
Die vollständige Auflistung der sonstigen Bestandteile siehe Abschnitt 6.1.
3. Darreichungsform
Weiße bis elfenbeinfarbene, geruchlose, torpedoförmige Zäpfchen zur rektalen Anwendung.
4. Klinische Angaben
4.1 Anwendungsgebiete
Symptomatische Behandlung leichter bis mäßig starker Schmerzen und/oder Fieber.
4.2 Dosierung, Art und Dauer der Anwendung
Die Dosierung richtet sich nach den Angaben in der nachfolgenden Tabelle.
Paracetamol wird in Abhängigkeit von Alter und Körpergewicht dosiert, in der Regel mit 10 bis 15 mg/kg Körpergewicht als Einzeldosis, bis maximal 60 mg/kg Körpergewicht als Tagesgesamtdosis.
Das jeweilige Dosierungsintervall richtet sich nach der Symptomatik und der maximalen Tagesgesamtdosis. Es sollte 6 Stunden nicht unterschreiten.
Bei Beschwerden die länger als 3 Tage anhalten, sollte ein Arzt aufgesucht werden.
|
Körpergewicht (Alter) |
Einzeldosis (entsprechende Paracetamoldosis) |
max. Tagesdosis (24 Std.) (entsprechende Paracetamoldosis) |
|
26 kg - 32 kg (8 bis 11 Jahre) |
1 Zäpfchen (entsprechend 500 mg Paracetamol) |
3 Zäpfchen (entsprechend 1.500 mg Paracetamol) |
|
33 kg - 43 kg (11 - 12 Jahre) |
1 Zäpfchen (entsprechend 500 mg Paracetamol) |
4 Zäpfchen (entsprechend 2.000 mg Paracetamol) |
|
Ab 43 kg (Kinder u. Jugendliche ab 12 Jahren und Erwachsene) |
1 -2 Zäpfchen (entsprechend 500 -1.000 mg Paracetamol) |
8 Zäpfchen (entsprechend 4.000 mg Paracetamol) |
Die in der Tabelle angegebene maximale Tagesdosis (24 Stunden) darf keinesfalls überschritten werden.
Art und Dauer der Anwendung
Die Zäpfchen werden möglichst nach dem Stuhlgang tief in den After eingelührt. Zur Verbesserung der Gleitfähigkeit eventuell Zäpfchen in der Hand erwärmen oder ganz kurz in warmes Wasser tauchen.
Besondere Patientengruppen
Leberinsuffizienz und leichte Niereninsuffizienz
Bei Patienten mit Leber- oder Nierenfunktionsstörungen sowie Gilbert-Syndrom muss die Dosis vermindert bzw. das Dosisintervall verlängert werden.
Schwere Niereninsuffizienz
Bei schwerer Niereninsuffizienz (Kreatinin-Clearance < 10 ml/min) muss ein Dosisintervall von mindestens 8 Stunden eingehalten werden.
Ältere Patienten
Es ist keine spezielle Dosisanpassung erforderlich.
Kinder und Jugendliche mit geringem Körpergewicht
Eine Anwendung von ben-u-ron 500 mg Zäpfchen bei Kindern unter 8 Jahren bzw. unter 26 kg Körpergewicht wird nicht empfohlen, da die Dosisstärke für diese Patientengruppe nicht geeignet ist. Es stehen jedoch für diese Patientengruppe geeignete Dosisstärken bzw. Darreichungsformen zur Verfügung.
4.3 Gegenanzeigen
Überempfindlichkeit gegen Paracetamol, Soja oder einen der sonstigen Bestandteile.
4.4 Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Um das Risiko einer Überdosierung zu vermeiden, sollte sichergestellt werden, dass gleichzeitig angewendete Medikamente kein Paracetamol enthalten.
Paracetamol sollte in folgenden Fällen mit besonderer Vorsicht (d.h. mit einem verlängerten Dosisintervall oder verminderter Dosis) und unter ärztlicher Kontrolle angewandt werden:
- Hepatozelluläre Insuffizienz
- Chronischer Alkoholmissbrauch
- Schwere Niereninsuffizienz (Kreatinin-Clearance < 10 ml/min (siehe Abschnitt 4.2))
- Gilbert-Syndrom (Meulengracht-Krankheit)
- Erkrankungen, die mit einem reduzierten Glutathionspiegel einhergehen können (ggf. Dosisanpassung z.B. bei Diabetes mellitus, HIV, Down-Syndrom, Tumoren)
Bei hohem Fieber, Anzeichen einer Sekundärinfektion oder Anhalten der Symptome über mehr als drei Tage, muss der Arzt konsultiert werden.
Allgemein sollen Paracetamol-haltige Arzneimittel ohne ärztlichen oder zahnärztlichen Rat nur wenige Tage und nicht in erhöhter Dosis angewendet werden.
Bei längerem hochdosierten, nicht bestimmungsgemäßen Gebrauch von Analgetika können Kopfschmerzen auftreten, die nicht durch erhöhte Dosen des Arzneimittels behandelt werden dürfen.
Ganz allgemein kann die gewohnheitsmäßige Einnahme von Schmerzmitteln, insbesondere bei Kombination mehrerer schmerzstillender Wirkstoffe zur dauerhaften Nierenschädigung mit dem Risiko eines Nierenversagens (Analgetika-Nephropathie) führen.
Bei abruptem Absetzen nach längerem hochdosierten, nicht bestimmungsgemäßen Gebrauch von Analgetika können Kopfschmerzen sowie Müdigkeit, Muskelschmerzen, Nervosität und vegetative Symptome auftreten. Die Absetzsymptomatik klingt innerhalb weniger Tage ab. Bis dahin soll die Wiedereinnahme von Schmerzmitteln unterbleiben und die erneute Einnahme nicht ohne ärztlichen Rat erfolgen.
4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
- Die Einnahme von Probenecid hemmt die Bindung von Paracetamol an Glucuronsäure und führt dadurch zu einer Reduzierung der Paracetamol-Clearance um ungefähr den Faktor 2. Bei gleichzeitiger Einnahme von Probenecid sollte die Paracetamoldosis verringert werden.
- Besondere Vorsicht ist bei der gleichzeitigen Einnahme von Arzneimitteln, die zu einer Enzyminduktion führen, sowie bei potenziell hepatotoxischen Substanzen geboten (siehe Abschnitt 4.9).
- Bei gleichzeitiger Anwendung von Paracetamol und AZT (Zidovudin) wird die Neigung zur Ausbildung einer Neutropenie verstärkt. Dieses Arzneimittel soll daher nur nach ärztlichem Anraten gleichzeitig mit AZT angewendet werden.
- Cholestyramin verringert die Aufnahme von Paracetamol.
Auswirkungen auf Laborwerte
Die Anwendung von Paracetamol kann die Harnsäurebestimmung mittels Phosphorwolframsäure sowie die Blutzuckerbestimmung mittels Glucose-Oxydase-Peroxydase beeinflussen.
4.6 Anwendung während Schwangerschaft und Stillzeit
Schwangerschaft
Epidemiologische Daten zur oralen Anwendung therapeutischer Dosen von Paracetamol geben keinen Hinweis auf mögliche unerwünschte Nebenwirkungen auf die Schwangerschaft oder die Gesundheit des Feten/Neugeborenen. Prospektive Daten zur Überdosierung während der Schwangerschaft zeigten keinen Anstieg des Risikos von Fehlbildungen. Reproduktionsstudien zur oralen Anwendung ergaben keinen Hinweis auf das Auftreten von Fehlbildungen oder Fetotoxizität.
Unter normalen Anwendungsbedingungen kann Paracetamol während der gesamten Schwangerschaft nach Abwägung des Nutzen-Risiko-Verhältnisses angewendet werden.
Während der Schwangerschaft sollte Paracetamol nicht über einen längeren Zeitraum, in höheren Dosen oder in Kombination mit anderen Arzneimitteln angewendet werden, da eine Sicherheit der Anwendung für diese Fälle nicht belegt ist.
Stillzeit
Nach der oralen Anwendung wird Paracetamol in geringen Mengen in die Muttermilch ausgeschieden. Bislang sind keine unerwünschten Wirkungen oder Nebenwirkungen während des Stillens bekannt. Paracetamol kann in der Stillzeit in therapeutischen Dosen verabreicht werden.
4.7 Auswirkungen auf die Verkehrstüchtigkeit und das Bedienen von Maschinen
Es sind keine negativen Auswirkungen zu erwarten.
4.8 Nebenwirkungen
Bei der Bewertung von Nebenwirkungen werden folgende Häufigkeiten zugrunde gelegt:
Sehr häufig (>1/10)
Häufig (>1/100 bis <1/10)
Gelegentlich (>1/1.000 bis <1/100)
Selten (>1/10.000 bis <1/1.000)
Sehr selten (<1/10.000)
Nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar)
Leber und Gallenerkrankungen Selten: Anstieg der Lebertransaminasen
Erkrankungen des Blutes und des Lymphsystems
Sehr selten: Veränderungen des Blutbildes wie Thrombozytopenie, Agranulozytose Erkrankungen des Immunsystems
Sehr selten: bei prädisponierten Personen Bronchospasmus (Analgetika-Asthma), Überempfindlichkeitsreaktionen von einfacher Hautrötung bis hin zu Urtikaria und anaphylaktischem Schock.
Erkrankungen der Haut und des Unterhautzellgewebes
Sehr selten wurden Fälle von schweren Hautreaktionen berichtet.
Andere mögliche Nebenwirkungen
Entölte Phospholipide aus Sojabohnen können sehr selten allergische Reaktionen hervorrufen.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-RisikoVerhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung über das nationale Meldesystem anzuzeigen: Bundesinstitut für Arzneimittel und Medizinprodukte, Abt. Pharmakovigilanz Kurt-Georg-Kiesinger Allee 3, D-53175 Bonn, www.bfarm.de Indem Sie Nebenwirkungen melden, können Sie dazu beitragen, dass mehr Informationen über die Sicherheit dieses Arzneimittels zur Verfügung gestellt werden.
4.9 Überdosierung
Symptome
Ein Intoxikationsrisiko besteht insbesondere bei älteren Menschen, kleinen Kindern, Personen mit Lebererkrankungen, chronischem Alkoholmissbrauch, chronischer Fehlernährung und bei gleichzeitiger Einnahme von Arzneimitteln, die zu einer Enzyminduktion führen. In diesen Fällen kann eine Überdosierung zum Tod führen.
In der Regel treten Symptome innerhalb von 24 Stunden auf: Übelkeit, Erbrechen, Anorexie, Blässe und Unterleibsschmerzen. Danach kann es zu einer Besserung des subjektiven Befindens kommen, es bleiben jedoch leichte Leibschmerzen als Hinweis auf eine Leberschädigung.
Eine Überdosierung mit ca. 6 g oder mehr Paracetamol als Einzeldosis bei Erwachsenen oder mit 140 mg/kg Körpergewicht als Einzeldosis bei Kindern führt zu Leberzellnekrosen, die zu einer totalen irreversiblen Nekrose und später zu hepatozellulärer Insuffizienz, metabolischer Azidose und Enzephalopathie führen können. Diese wiederum können zu Koma, auch mit tödlichem Ausgang, führen. Gleichzeitig wurden erhöhte Konzentrationen der Lebertransaminasen (AST, ALT), Laktatdehydrogenase und des Bilirubins in Kombination mit einer erhöhten Prothrombinzeit beobachtet, die 12 bis 48 Stunden nach der Anwendung auftreten können. Klinische Symptome der Leberschäden werden in der Regel nach 2 Tagen sichtbar und erreichen nach 4 bis 6 Tagen ein Maximum.
Auch wenn keine schweren Leberschäden vorliegen, kann es zu akutem Nierenversagen mit akuter Tubulusnekrose kommen. Zu anderen, leberunabhängigen Symptomen, die nach einer Überdosierung mit Paracetamol beobachtet wurden, zählen Myokardanomalien und Pankreatitis.
Therapiemaßnahmen bei Überdosierung
Bereits bei Verdacht auf Intoxikation mit Paracetamol ist
in den ersten 10 Stunden die intravenöse Gabe von SH-Gruppen-Donatoren wie z. B. N-Acetylcystein sinnvoll. N-Acetylcystein kann aber auch nach 10 und bis zu 48 Stunden noch einen gewissen Schutz bieten. In diesem Fall erfolgt eine längerfristige Einnahme. Durch Dialyse kann die Plasmakonzentration von Paracetamol abgesenkt werden. Bestimmungen der Plasmakonzentration von Paracetamol sind empfehlenswert.
Die weiteren Therapiemöglichkeiten zur Behandlung einer Intoxikation mit Paracetamol richten sich nach Ausmaß, Stadium und klinischen Symptomen entsprechend den üblichen Maßnahmen in der Intensivmedizin.
5. Pharmakologische Eigenschaften
Pharmakotherapeutische Gruppe: Analgetika und Antipyretika, Anilide ATC-Code: N02BE01
5.1 Pharmakody na mische Eigenschaften
Der analgetische und antipyretische Wirkungsmechanismus von Paracetamol ist nicht eindeutig geklärt. Eine zentrale und periphere Wirkung ist wahrscheinlich. Nachgewiesen ist eine ausgeprägte Hemmung der cerebralen Prostaglandinsynthese, während die periphere Prostaglandinsynthese nur schwach gehemmt wird. Ferner hemmt Paracetamol den Effekt endogener Pyrogene auf das hypothalamische Temperaturregulationszentrum.
5.2 Pharmakokinetische Eigenschaften
Resorption
Nach oraler Gabe wird Paracetamol rasch und vollständig resorbiert. Maximale Plasmakonzentrationen werden 30 bis 60 Minuten nach der Einnahme erreicht.
Nach rektaler Gabe wird Paracetamol zu 68 - 88 % resorbiert; maximale Plasmakonzentrationen werden erst nach 3 - 4 Stunden erreicht.
Bioverfügbarkeit
Eine im Jahr 1992 durchgeführte Bioverfügbarkeitsuntersuchung an 17 Probanden ergab für das Zäpfchen mit 500 mg als Testpräparat im Vergleich zur Tablette mit 500 mg als Referenzpräparat:
|
Testpräparat |
Referenzpräparat | |
|
Maximale Plasmakonzentration Cmax [mg/l]: |
3,81 ± 1,42 |
7,31 ± 1,36 |
|
Zeitpunkt der maximalen Plasmakonzentration Wx[h]: |
2,0 ± 0,46 |
0,56 ± 0,34 |
|
Fläche unter der Konzentrations Zeit-Kurve AUCO - t [mg/1 x h]: |
19,71 ± 7,91 |
21,92 ± 7,31 |
Angabe der W erte als Mittelwert und Streubreite
Mittlere Plasmaspiegelverläufe im Vergleich zu einem Referenzpräparat in einem Kon-zentrations-Zeit-Diagramm:
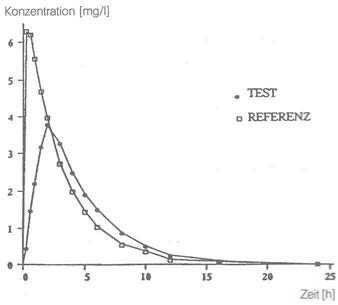
Die relative Bioverfügbarkeit für ben-u-ron® 500 mg Suppositorien beträgt 90,8 % bezogen auf die Tablettenformulierung.
Verteilung
Paracetamol verteilt sich rasch in allen Geweben. Blut-, Plasma- und Speichelkonzentrationen sind vergleichbar. Die Plasmaproteinbindung ist gering.
Stoffwechsel
Paracetamol wird vorwiegend in der Leber auf hauptsächlich zwei Wegen metabolisiert: Konjugation mit Glucuronsäure und Schwefelsäure. Bei Dosen, die die therapeutische Dosis übersteigen, ist der zuletzt genannte Weg rasch gesättigt. Ein geringer Teil der Me-tabolisierung erfolgt über den Katalysator Cytochrom P 450 (hauptsächlich CYP2E1) und führt zur Bildung des Metaboliten N-Acetyl-p-benzochinonimin, der normalerweise rasch durch Glutathion entgiftet und durch Cystein und Mercaptursäure gebunden wird. Im Falle einer massiven Intoxikation ist die Menge dieses toxischen Metaboliten erhöht.
Elimination
Die Ausscheidung erfolgt vorwiegend im Urin. 90% der aufgenommenen Menge werden innerhalb von 24 Stunden vorwiegend als Glucuronide (60 bis 80%) und Sulphatkonjuga-te (20 bis 30%) über die Nieren ausgeschieden. Weniger als 5% werden in unveränderter Form ausgeschieden.
Die Eliminationshalbwertzeit beträgt in etwa zwei Stunden. Bei Leber- und Nierenfunktionsstörungen, nach Überdosierungen sowie bei Neugeborenen ist die Halbwertszeit verlängert. Das Maximum der Wirkung und die durchschnittliche Wirkdauer (4 - 6 Stunden) korrelieren in etwa mit der Plasmakonzentration.
Niereninsuffizienz
Bei schwerer Niereninsuffizienz (Kreatinin-Clearance < 10 ml/min) ist die Ausscheidung von Paracetamol und seinen Metaboliten verzögert.
Ältere Patienten
Die Fähigkeit zur Konjugation ist unverändert.
5.3 Präklinische Daten zur Sicherheit
In Tierversuchen zur akuten, subchronischen und chronischen Toxizität von Paracetamol, an Ratte und Maus, wurden gastrointestinale Läsionen, Veränderungen im Blutbild, dege-nerative Veränderungen des Leber- und Nierenparenchyms sowie Nekrosen beobachtet. Der Grund für diese Veränderungen ist einerseits im Wirkungsmechanismus und andererseits im Metabolismus von Paracetamol zu suchen. Diejenigen Metaboliten, die vermutlich Ursache der toxischen Wirkung und der daraus folgenden Veränderungen an Organen sind, wurden auch beim Menschen gefunden. Während einer Langzeitanwendung (das heißt 1 Jahr) im Bereich maximaler therapeutischer Dosen wurden auch sehr seltene Fälle einer reversiblen chronischen aggressiven Hepatitis beobachtet. Bei subtoxischen Dosen können nach dreiwöchiger Einnahme Intoxikationssymptome auftreten. Daher sollte Paracetamol nicht über längere Zeit und nicht in höheren Dosen eingenommen werden.
Umfangreiche Untersuchungen ergaben keine Evidenz für ein relevantes genotoxisches Risiko von Paracetamol im therapeutischen, das heißt nicht-toxischen Dosisbereich.
Aus Langzeituntersuchungen an Ratten und Mäusen liegen keine Hinweise auf relevante tumorigene Effekte in nicht-hepatotoxischen Dosierungen von Paracetamol vor.
Paracetamol passiert die Plazenta.
Aus Tierstudien und den bisherigen Erfahrungen an Menschen ergeben sich keine Hinweise auf Fruchtschädigungen.
6. Pharmazeutische Angaben 6.1 Liste der sonstigen Bestandteile
Hartfett, entölte Phospholipide aus Sojabohnen
6.2 Inkompatibilitäten
Nicht zutreffend.
6.3 Dauer der Haltbarkeit
Die Dauer der Haltbarkeit beträgt 5 Jahre.
Dieses Arzneimittel soll nach Ablauf des Verfalldatums nicht mehr angewendet werden.
6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Nicht über 25 °C lagern.
6.5 Art und Inhalt des Behältnisses
ben-u-ron® 500 mg
Aluminium Blisterstreifen mit 5 Zäpfchen. OP mit 10 Zäpfchen (N1)
AP mit 10 x 10 Zäpfchen
6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung
Keine besonderen Anforderungen.
7. Inhaber der Zulassung
bene-Arzneimittel GmbH Herterichstraße 1 81479 München S Postfach 710269 81452 München Telefon: 0 89 / 7 49 87-0 Telefax: 0 89 / 7 49 87- 142 contact@b ene- arzne i mittel. de
8. Zulassungsnummer
6012055.02.00
9. Datum der Erteilung der Zulassung / Verlängerung der Zulassung 23.10.1996 / 01.12.2006
10. Stand der Information
März 2014
11. Verkaufsabgrenzung
Apothekenpflichtig
10 / 10