Beriate 2000
CSL Behring
Beriate® 250/500/1000/2000
1. BEZEICHNUNG DES ARZNEIMITTELS
Beriate® 250 I.E., Pulver und Lösungsmittel zur Herstellung einer Injektions- / Infusionslösung
Beriate® 500 I.E., Pulver und Lösungsmittel zur Herstellung einer Injektions- / Infusionslösung
Beriate® 1000 I.E., Pulver und Lösungsmittel zur Herstellung einer Injektions- / Infusionslösung
Beriate® 2000 I.E., Pulver und Lösungsmittel zur Herstellung einer Injektions- / Infusionslösung
2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
Eine Flasche enthält nominal:
250/500/1000/2000 I.E. humanen Blutgerinnungsfaktor VIII (FVIII).
Nach Rekonstitution mit 2,5/5/10 ml enthält Beriate 250/500/1000 100 I.E./ml Gerinnungsfaktor VIII vom Menschen. Beriate 2000 wird mit 10 ml Wasser für Injektionszwecke rekonstituiert und enthält ca. 200 I.E./ml Gerinnungsfaktor VIII vom Menschen.
Die Aktivität (I.E.) wird mittels chromogenem Test gemäß Europäischem Arzneibuch bestimmt. Die mittlere spezifische Aktivität von Beriate beträgt ca. 270 I.E./mg Protein.
Hergestellt aus Plasma humaner Spender.
Sonstiger Bestandteil mit bekannter Wirkung:
Natrium etwa 100 mmol/l (2,3 mg/ml).
Die vollständige Auflistung der sonstigen Bestandteile siehe Abschnitt 6.1.
3. DARREICHUNGSFORM
Pulver und Lösungsmittel zur Herstellung einer Injektions- / Infusionslösung.
Weißes Pulver und klares, farbloses Lösungsmittel zur Herstellung einer Injektions- oder Infusionslösung.
4. KLINISCHE ANGABEN
4.1 Anwendungsgebiete
Therapie und Prophylaxe von Blutungen bei Patienten mit Hämophilie A (kongenitaler Faktor-VIII-Mangel).
Dieses Produkt kann in der Behandlung des erworbenen Faktor-VIII-Mangels eingesetzt werden.
4.2 Dosierung und Art der Anwendung
Die Therapie soll unter der Aufsicht eines in der Hämophilie-Behandlung erfahrenen Arztes erfolgen.
Dosierung
Dosierung und Dauer der Substitutionstherapie richten sich nach dem Schweregrad des Faktor-VIII-Mangels, nach Ort und Ausmaß der Blutung und nach dem klinischen Zustand des Patienten.
Die Menge des verabreichten Faktor VIII wird in Internationalen Einheiten (I.E.) angegeben, die dem gegenwärtigen WHO-Standard für Faktor-VIII-Produkte entsprechen. Die Faktor-VIII-Aktivität im Plasma wird entweder als Prozentsatz (bezogen auf das normale Humanplasma) oder in I.E. (bezogen auf den Internationalen Standard für Faktor VIII im Plasma) angegeben.
Eine I.E. Faktor-VIII-Aktivität entspricht dem Faktor-VIII-Gehalt von 1 ml normalem Humanplasma.
Bedarfsbehandlung:
Die Berechnung der benötigten Dosis an Faktor VIII basiert auf dem empirischen Ergebnis, dass 1 I.E. Faktor VIII pro kg Körpergewicht die Faktor-VIII-Aktivität im Plasma um ca. 2 % (2 I.E./dl) der normalen Aktivität anhebt. Die benötigte Dosis wird nach folgender Formel berechnet:
Erforderliche Einheiten = Körpergewicht [kg] x gewünschter Faktor-VIII-Anstieg [% oder I.E./dl] x 0,5.
Dosierung und die Häufigkeit der Anwendung sollen sich stets an der klinischen Wirksamkeit im Einzelfall orientieren.
Bei den folgenden Blutungsereignissen soll die Faktor-VIII-Aktivität im Plasma (in % der Norm oder I.E./dl) im entsprechenden Zeitraum nicht unterschritten werden. Die folgende Tabelle dient als Empfehlung für die Dosierung bei Blutungsereignissen und chirurgischen Eingriffen:
|
Schweregrad der Blutung/Art des chirurgischen Eingriffs |
Benötigter Faktor-VIII-Spiegel (% oder I.E./dl) |
Häufigkeit der Dosierung (Stunden)/Dauer der Behandlung (Tage) |
|
Blutung | ||
|
Beginnende Hämarthrosen, Muskelblutungen oder Blutungen in der Mundhöhle |
20-40 |
Wiederholung der Infusion alle 12 bis 24 Stunden. Mind. 1 Tag, bis Beendigung des Blutungsereignisses (durch Schmerz angezeigt) oder bis Abschluss der Wundheilung. |
|
Ausgedehntere Hämarthrosen, Muskelblutungen oder Hämatome |
30-60 |
Wiederholung der Infusion alle 12 bis 24 Stunden für 3 bis 4 Tage, oder länger, bis zur Beseitigung des Schmerzzustandes und der akuten Bewegungseinschränkung. |
|
Lebensbedrohliche Blutungen |
60-100 |
Wiederholung der Infusion alle 8 bis 24 Stunden bis zur Aufhebung des lebensbedrohlichen Zustandes. |
|
Chirurgische Eingriffe | ||
|
Kleinere Eingriffe einschließlich Zahnextraktion |
30-60 |
Alle 24 Stunden, mind. 1 Tag, bis Abschluss der Wundheilung. |
|
Größere Eingriffe |
80-100 (prä- und postoperativ) |
Wiederholung der Infusion alle 8 bis 24 Stunden bis zum adäquaten Abschluss der Wundheilung, dann Behandlung für mind. weitere 7 Tage zur Erhaltung einer Faktor-VIII-Aktivität von 30 % bis 60 % (I.E./dl). |
Prophylaxe:
Bei der Langzeitprophylaxe von Blutungen bei Patienten mit schwerer Hämophilie A betragen die üblichen Dosen 20 bis 40 I.E. Faktor VIII pro kg Körpergewicht in Intervallen von 2 bis 3 Tagen. In manchen Fällen, besonders bei jüngeren Patienten, können kürzere Dosierungsintervalle oder höhere Dosen notwendig sein.
Während der Behandlung sollten die Faktor-VIII-Spiegel entsprechend bestimmt werden, um die zu verabreichende Dosis und die Häufigkeit wiederholter Infusionen zu steuern. Vor allem bei größeren chirurgischen Eingriffen ist eine genaue gerinnungsanalytische Überwachung (Faktor-VIII-Aktivität im Plasma) der Substitutionstherapie unerlässlich. Einzelne Patienten können in ihrem Ansprechen auf Faktor VIII variieren, wobei unterschiedliche Werte der In-vivo-Recovery erreicht und unterschiedliche Halbwertszeiten gesehen werden.
Die Patienten sollen bezüglich einer Entwicklung von Hemmkörpern gegen Faktor VIII überwacht werden. Siehe auch Abschnitt 4.4.
Zuvor unbehandelte Patienten:
Die Sicherheit und Wirksamkeit von Beriate bei zuvor unbehandelten Patienten wurde bisher nicht untersucht.
Kinder und Jugendliche:
Die Dosierung bei Kindern richtet sich nach dem Körpergewicht und deshalb generell nach den gleichen Richtlinien wie für Erwachsene. Die Häufigkeit der Anwendung soll sich stets an der klinischen Wirksamkeit im Einzelfall orientieren. Erfahrungen in der Behandlung von Kindern < 6 Jahre liegen vor (siehe auch Abschnitt 5.1).
Art der Anwendung
Zur intravenösen Anwendung.
Für Informationen zur Rekonstitution des Produktes vor der Anwendung, siehe Abschnitt 6.6.
Die rekonstituierte Lösung soll vor der Anwendung auf Raum- oder Körpertemperatur angewärmt und langsam intravenös mit einer vom Patienten als angenehm empfundenen Geschwindigkeit injiziert oder infundiert werden. Die Injektions- / Infusionsgeschwindigkeit sollte nicht mehr als 2 ml pro Minute betragen.
Der Patient ist auf jegliche Sofortreaktionen zu beobachten. Wenn eine Reaktion erfolgt, die mit der Verabreichung von Beriate in Zusammenhang gebracht werden könnte, soll - in Abhängigkeit vom klinischen Zustand des Patienten - die Infusionsgeschwindigkeit gesenkt bzw. die Infusion abgebrochen werden (siehe auch Abschnitt 4.4).
4.3 Gegenanzeigen
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Überempfindlichkeit
Allergieartige Überempfindlichkeitsreaktionen sind möglich. Falls Symptome einer Überempfindlichkeit auftreten, sollte den Patienten geraten werden, die Anwendung des Produkts sofort zu unterbrechen und ihren Arzt aufzusuchen. Patienten sollen über Frühzeichen von Überempfindlichkeitsreaktionen informiert werden, einschließlich quaddelartiger Hautausschlag, generalisierte Nesselsucht, Engegefühl in der Brust, Stridor, Hypotonie und Anaphylaxie.
Bei einem Schock sollen die aktuellen medizinischen Richtlinien zur Schockbehandlung beachtet werden.
Inhibitoren
Die Bildung neutralisierender Antikörper (Hemmkörper) gegen Faktor VIII ist eine bekannte Komplikation bei der Behandlung von Hämophilie A-Patienten. Diese Hemmkörper sind gewöhnlich IgG Immunglobuline, die sich gegen die Faktor-VIII-Gerinnungsaktivität richten. Sie werden mittels modifiziertem Test in Bethesda-Einheiten (BE) pro ml Plasma quantifiziert. Das Risiko der Bildung von Hemmkörpern korreliert mit der Aufnahme von Faktor VIII, wobei das Risiko in den ersten 20 Tagen nach Exposition am höchsten ist. In seltenen Fällen entwickeln sich Hemmkörper nach den ersten 100 Tagen.
Fälle von wiederkehrenden Inhibitoren (mit niedrigem Inhibitortiter/Inhibitorspiegel) wurden nach Umstellen von einem FVIII-Produkt zu einem anderen bei zuvor behandelten Patienten mit mehr als 100 Expositionstagen beobachtet, die eine Inhibitorentwicklung in der Vorgeschichte haben. Deshalb wird empfohlen, alle Patienten nach jedem Wechsel des Produktes sorgfältig auf das Auftreten von Inhibitoren zu beobachten.
Grundsätzlich sollten alle Patienten, die mit Gerinnungsfaktor VIII vom Menschen behandelt wurden, durch geeignete klinische Beobachtung und Labortests sorgfältig bezüglich der Entwicklung von Hemmkörpern beobachtet werden. Wenn der erwartete Plasmaspiegel der FVIII-Aktivität im Plasma nicht erreicht wird, oder wenn die Blutung nicht mit einer angemessenen Dosis kontrolliert wird, soll ein Test zum Nachweis von FVIII-Inhibitoren durchgeführt werden. Bei Patienten mit hohen Inhibitorspiegeln kann die FVIII-Behandlung unwirksam sein und es sollten andere Behandlungsmöglichkeiten erwogen werden. Die Behandlung solcher Patienten soll von Ärzten, die in der Therapie von Hämophilie A-Patienten und Patienten mit FVIII-Inhibitoren erfahren sind, durchgeführt werden. Siehe auch Abschnitt 4.8.
Beriate enthält bis zu 28 mg Natrium pro 1000 I.E. Dies sollte bei Patienten, die eine salzarme Diät einhalten müssen, berücksichtigt werden.
Virussicherheit
Standardmethoden zur Vermeidung von Infektionskrankheiten, die im Rahmen der Anwendung von aus menschlichem Blut oder Plasma hergestellten Arzneimitteln auftreten können, umfassen die Auswahl der Spender, die Prüfung individueller Spenden und der Plasmapools auf spezifische Marker für Infektionen sowie die Einbeziehung effektiver Herstellungsschritte zur Inaktivierung/Eliminierung von Viren. Trotz dieser Maßnahmen kann die Möglichkeit der Übertragung von Erregern bei der Anwendung von aus menschlichem Blut oder Plasma hergestellten Arzneimitteln nicht vollständig ausgeschlossen werden. Dies gilt auch für bisher unbekannte Viren und andere Pathogene.
Die getroffenen Maßnahmen werden als wirksam angesehen für umhüllte Viren, wie z.B. das humane Immundefizienzvirus (HIV), das Hepatitis B-Virus (HBV) und das Hepatitis C-Virus (HCV) sowie für die nicht-umhüllten Viren Hepatitis A (HAV) und Parvovirus B19.
Für Patienten, die regelmäßig Präparate aus menschlichem Blut oder Plasma einschließlich Faktor VIII Präparate erhalten, wird grundsätzlich eine Impfung gegen Hepatitis A und Hepatitis B empfohlen.
Es wird auf die Dokumentationspflicht gemäß Transfusionsgesetz hingewiesen.
Kinder und Jugendliche
Die aufgeführten Warnhinweise und Vorsichtsmaßnahmen gelten für Erwachsene und für Kinder.
4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Es sind bisher keine Wechselwirkungen von FVIII mit anderen Arzneimitteln untersucht worden.
4.6 Fertilität, Schwangerschaft und Stillzeit
Reproduktionsstudien am Tier wurden mit Faktor VIII nicht durchgeführt.
Schwangerschaft und Stillzeit
Aufgrund des seltenen Vorkommens der Hämophilie A bei Frauen liegen keine Erfahrungen über die Anwendung von Faktor VIII während der Schwangerschaft und Stillzeit vor.
Daher soll Faktor VIII in der Schwangerschaft und Stillzeit nur bei klarer Indikationsstellung angewendet werden.
Fertilität
Es liegen keine Daten zur Fertilität vor.
4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Beriate hat keinen Einfluss auf die Verkehrstüchtigkeit und Fähigkeit zum Bedienen von Maschinen.
4.8 Nebenwirkungen
Zusammenfassung des Sicherheitsprofiles
Überempfindlichkeitsreaktionen oder allergische Reaktionen (die auch Angioödem, Brennen und Stechen an der Infusionsstelle, Schüttelfrost, Hautrötung mit Hitzegefühl, generalisierte Urtikaria, Kopfschmerzen, Nesselausschlag, Hypotonie, Lethargie, Übelkeit, Unruhe, Tachykardie, Engegefühl in der Brust, Zittern, Erbrechen und Stridor mit einschließen können) wurden sehr selten beobachtet und können sich in manchen Fällen zu schwerer Anaphylaxie (einschließlich Schock) entwickeln.
Patienten mit Hämophilie A können sehr selten neutralisierende Antikörper (Hemmkörper) gegen Faktor VIII entwickeln. Wenn solche Hemmkörper auftreten, manifestiert sich der Zustand als unzureichende klinische Antwort. In solchen Fällen wird empfohlen, ein spezialisiertes Hämophilie-Zentrum aufzusuchen.
Tabellarische Aufstellung der Nebenwirkungen
Die im folgenden genannten Nebenwirkungen beruhen auf Analysen von postmarketing Daten sowie der wissenschaftlichen Literatur.
Die unten aufgeführte Tabelle entspricht der MedDRA Systemorganklassifikation.
Die Häufigkeiten wurden entsprechend der nachfolgenden Konventionen berechnet: sehr häufig (>1/10); häufig (> 1/100 und < 1/10); gelegentlich (> 1/1000 und < 1/100); selten (> 1/10.000 und < 1/1000); sehr selten (< 1/10.000); nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
|
MedDRA Systemorganklasse |
Nebenwirkung |
Häufigkeit |
|
Erkrankungen des Blutes und des Lymphsystems |
FVIII Inhibitoren |
Sehr selten |
|
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort |
Fieber |
Sehr selten |
|
Erkrankungen des Immunsystems |
Überempfindlichkeitsreaktionen (allergische Reaktionen) |
Sehr selten |
Informationen zum Infektionsrisiko siehe Abschnitt 4.4 . Kinder und Jugendliche
Es wird erwartet, dass die Häufigkeit, Art und Schwere der Nebenwirkungen bei Kindern denen bei Erwachsenen entspricht.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-RisikoVerhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel, Paul-Ehrlich-Institut, Paul-Ehrlich-Str. 51-59, 63225 Langen, Tel: +49 6103 77 0, Fax: +49 6103 77 1234, Website: www.pei.de anzuzeigen.
4.9 Überdosierung
Bisher wurden keine Symptome von Überdosierung mit Gerinnungsfaktor VIII vom Menschen beobachtet.
5. PHARMAKOLOGISCHE EIGENSCHAFTEN
5.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe Antihämorrhagika: Blutgerinnungsfaktoren.
ATC-Code: B02BD02
Der Faktor-VIII-/von-Willebrand-Faktor-Komplex besteht aus 2 Untereinheiten (Faktor VIII und von-Willebrand-Faktor), die unterschiedliche physiologische Wirkungen besitzen.
Bei der Behandlung hämophiler Patienten, bindet Faktor VIII an von-Willebrand-Faktor im Blutkreislauf des Patienten.
Aktivierter Faktor VIII agiert als Kofaktor für den aktivierten Faktor IX und beschleunigt so die Umwandlung von Faktor X in aktivierten Faktor X. Aktivierter Faktor X wandelt Prothrombin in Thrombin um. Thrombin wandelt dann Fibrinogen in Fibrin um, und ein Gerinnsel kann gebildet werden. Hämophilie A ist eine geschlechtsgebunden vererbte Störung der Blutgerinnung, die auf einem Mangel an Faktor VIII:C beruht. Als Folge davon treten spontane oder verletzungsbedingte oder bei chirurgischen Eingriffen hervorgerufene Blutungen in Gelenken, Muskeln oder inneren Organen auf. Durch Substitutionstherapie werden die Faktor-VIII-Plasmaspiegel angehoben und dadurch eine vorübergehende Korrektur des Faktor-Mangels sowie eine Behebung der Blutungsneigungen herbeigeführt.
Zusätzlich zu seiner Funktion als Faktor VIII stabilisierendes Protein vermittelt von Willebrand Faktor die Anlagerung der Thrombozyten an den verletzten Gefäßendothelien und spielt die Hauptrolle bei der Thrombozytenaggregation.
Daten zur Behandlung von 16 Kindern jünger als 6 Jahren liegen vor. Die vorliegenden Ergebnisse zur klinischen Wirksamkeit und Sicherheit waren in Übereinstimmung mit den Daten bei älteren Patienten.
5.2 Pharmakokinetische Eigenschaften
Nach intravenöser Anwendung nimmt die Faktor-VIII-Aktivität mono- oder biexponentiell ab. Die terminale Halbwertszeit variiert zwischen 5 und 22 Stunden, mit einem Mittelwert von ca. 12 Stunden. Der Anstieg der Faktor-VIII-Aktivität nach Gabe von 1 I.E. Faktor VIII pro kg Körpergewicht (inkrementelle Wiederfindungsrate) betrug ungefähr 2%; die intraindividuelle Variabilität lag zwischen 1,5 bis 3%. Die mittlere Verweildauer war 17 Stunden (Standardabweichung 5,5 Stunden). Die mittlere Fläche unter der Konzentrations-Zeitkurve (bis zum letzten Messpunkt) war 0,4 h x kg/ml (Standardabweichung 0,2), die mittlere Clearance 3 ml/h/kg (Standardabweichung 1,5 ml/h/kg).
Kinder und Jugendliche
Es liegen begrenzte pharmakokinetische Daten für Kinder vor.
5.3 Präklinische Daten zur Sicherheit
Allgemeine Toxizität
Toxizitätsstudien mit wiederholten Dosen wurden wegen der Antikörperbildung gegen heterologes Protein nicht durchgeführt.
Sogar das Mehrfache der empfohlenen menschlichen Dosis pro kg Körpergewicht zeigte keine toxischen Wirkungen bei Labortieren.
Die Prüfungen des hitzebehandelten Faktor-VIII-Präparates mit polyklonalen präzipitierenden Antikörpern (Kaninchen) im Ouchterlony-Test und in dem passiven kutanen Anaphylaxie-Test am Meerschweinchen zeigten im Vergleich zu unbehandeltem Protein keine veränderten immunologischen Reaktionen.
Mutagenität
Da die klinische Erfahrung keinen Hinweis auf onkogene und mutagene Wirkungen des Gerinnungsfaktors VIII vom Menschen gibt, werden Tierversuche, besonders bei heterologen Spezies, als nicht sinnvoll erachtet.
6. PHARMAZEUTISCHE ANGABEN 6.1 Liste der sonstigen Bestandteile
Glycin, Calciumchlorid, Natriumhydroxid (in geringen Mengen) zur Einstellung des pH-Wertes, Saccharose, Natriumchlorid
Beigefügtes Lösungsmittel: Wasser für Injektionszwecke 2,5/5/10 ml
6.2 Inkompatibilitäten
Dieses Arzneimittel darf, außer mit den unter Abschnitt 6.1 aufgeführten Lösungsmitteln, nicht mit anderen Arzneimitteln, Lösungs- oder Verdünnungsmitteln vermischt werden.
6.3 Dauer der Haltbarkeit
3 Jahre.
Die physiko-chemische Stabilität des rekonstituierten Präparates wurde für 8 Stunden bei 25 °C belegt. Aus mikrobiologischer Sicht sollte das Präparat sofort verbraucht werden. Falls es nicht sofort angewendet wird, soll eine Aufbewahrung 8 Stunden bei Raumtemperatur nicht überschreiten.
6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Im Kühlschrank lagern (2°C - 8°C). Nicht einfrieren. Behältnis in der geschlossenen Faltschachtel aufbewahren, um den Inhalt vor Licht zu schützen.
Beriate kann innerhalb der Haltbarkeitsdauer für einen Zeitraum von insgesamt 1 Monat bei bis zu 25 °C gelagert werden. Die einzelnen Zeitabschnitte der Raumtemperaturlagerung sollten dokumentiert werden, um den Gesamtzeitraum von 1 Monat nicht zu überschreiten. Die Flaschen nicht direkter Hitze aussetzen. Die Flaschen dürfen nicht über Körpertemperatur (37°C) erhitzt werden.
Aufbewahrungsbedingungen nach Rekonstitution des Arzneimittels, siehe Abschnitt 6.3.
6.5 Art und Inhalt der Behältnisse
Eine Packung mit 250 I.E. enthält:
1 Flasche mit Trockensubstanz 1 Flasche mit 2,5 ml Wasser für Injektionszwecke Eine Packung mit Zubehör enthält:
1 Filter Transfer Set 20/20 1 Einmalspritze (5 ml)
1 Venenpunktionsbesteck
2 Alkoholtupfer
1 nicht steriles Pflaster
Eine Packung mit 500 I.E. enthält:
1 Flasche mit Trockensubstanz 1 Flasche mit 5 ml Wasser für Injektionszwecke Eine Packung mit Zubehör enthält:
1 Filter Transfer Set 20/20 1 Einmalspritze (5 ml)
1 Venenpunktionsbesteck
2 Alkoholtupfer
1 nicht steriles Pflaster
Eine Packung mit 1000 I.E. enthält:
1 Flasche mit Trockensubstanz 1 Flasche mit 10 ml Wasser für Injektionszwecke Eine Packung mit Zubehör enthält:
1 Filter Transfer Set 20/20 1 Einmalspritze (10 ml)
1 Venenpunktionsbesteck
2 Alkoholtupfer
1 nicht steriles Pflaster
Eine Packung mit 2000 I.E. enthält:
1 Flasche mit Trockensubstanz 1 Flasche mit 10 ml Wasser für Injektionszwecke Eine Packung mit Zubehör enthält:
1 Filter Transfer Set 20/20 1 Einmalspritze (10 ml)
1 Venenpunktionsbesteck
2 Alkoholtupfer
1 nicht steriles Pflaster
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung
Art der Anwendung
Allgemeine Hinweise
- Die Lösung sollte klar oder leicht opaleszent sein. Rekonstituiertes Produkt sollte nach der Filtration/dem Aufziehen der Lösung in die Spritze (siehe unten) und vor der Anwendung auf Partikel und Verfärbungen visuell überprüft werden.
Trübe Lösungen oder Lösungen mit Rückständen (Niederschlägen/Partikeln) sind nicht zu verwenden.
- Zubereitung und Entnahme müssen unter aseptischen Bedingungen erfolgen.
CSL Behring
Beriate® 250/500/1000/2000 Zubereitung
Erwärmen Sie das Lösungsmittel auf Raumtemperatur. Vor dem Öffnen der Mix2Vial Packung die Flip-Off-Kappen der Lösungsmittel- und Produktflaschen entfernen und die Stopfen mit einer antiseptischen Lösung behandeln und anschließend trocknen lassen.

1
2
3
1. Entfernen Sie das Deckpapier von der Mix2Vial Packung. Das Mix2Vial nicht aus dem Blister entnehmen!
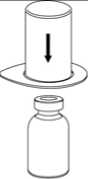
2. Die Lösungsmittelflasche auf eine ebene, saubere Fläche stellen und festhalten. Das Mix2Vial Set mit dem Blister greifen und den Dorn des blauen Adapters senkrecht in den Stopfen der Lösungsmittelflasche einstechen.
3. Vorsichtig die Verpackung vom Mix2Vial Set entfernen, indem man den Blister am Siegelrand fasst und ihn senkrecht nach oben abzieht. Dabei ist darauf zu achten, dass nur der Blister und nicht das Mix2Vial entfernt wird.

4
4. Die Produktflasche auf eine feste Unterlage stellen. Die Lösungsmittelflasche mit dem aufgesetzten Mix2Vial Set herumdrehen und den Dorn des transparenten Adapters senkrecht in den Stopfen der Produktflasche einstechen. Das Lösungsmittel läuft automatisch in die Produktflasche über.
Q
HL

6
5. Mit der einen Hand die Produktseite und mit der anderen Hand die Lösungsmittelseite des Mix2Vial greifen und das Set vorsichtig gegen den Uhrzeigersinn auseinander schrauben. Entsorgen Sie die Lösungsmittelflasche mit dem blauen Mix2Vial Adapter.
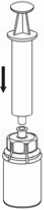
6. Die Produktflasche mit dem transparenten Adapter vorsichtig schwenken, bis das Produkt vollständig gelöst ist. Nicht schütteln.
7
7. Luft in eine leere, sterile Spritze aufziehen. Die Produktflasche aufrecht halten, die Spritze mit dem Luer Lock Anschluss des Mix2Vial Set verbinden indem man sie im Uhrzeigersinn aufschraubt, und die Luft in die Produktflasche injiziert.
Aufziehen der Lösung in die Spritze und Anwendung
|
i |
k |
8. Den Stempel der Spritze gedrückt halten, das gesamte System herumdrehen und die Lösung durch langsames Zurückziehen der Kolbenstange in die Spritze aufziehen. |
|
9. Nachdem die Lösung vollständig in die | ||
|
> |
Spritze überführt ist, den Spritzenzylinder fassen (dabei die Kolbenstange in ihrer | |
|
* |
i |
Position halten) und die Spritze vom transparenten Mix2Vial Adapter gegen den Uhrzeigersinn abdrehen. |
|
L |
, |
Zur Injektion von Beriate werden Kunststoff-Einmalspritzen empfohlen, da geschliffene Oberflächen von Glasspritzen zur Verklebung bei derartigen Lösungen neigen.
Die Lösung langsam intravenös verabreichen (siehe Abschnitt 4.2). Sorgfältig darauf achten, dass kein Blut in die mit Produkt gefüllte Spritze gelangt. Verwenden Sie hierzu das beiliegende Venenpunktionsbesteck, führen Sie die Nadel in eine Vene. Lassen Sie das Blut bis zum Ende des Schlauchs fließen. Befestigen Sie die Spritze an dem Luer-Ende des Venenpunktionsbestecks.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
CSL Behring
Beriate® 250/500/1000/2000
7. INHABER DER ZULASSUNG
CSL Behring GmbH
- Emil-von-Behring-Str. 76 35041 Marburg
- Verkauf Deutschland Philipp-Reis-Str. 2 65795 Hattersheim Tel.: (069) 305 - 8 44 37 Fax: (069) 305 - 1 71 29
8. ZULASSUNGSNUMMERN
|
Beriate® 250 |
Zul.-Nr.: |
10505a/98-1 |
|
Beriate® 500 |
Zul.-Nr.: |
10505a/98-2 |
|
Beriate® 1000 |
Zul.-Nr.: |
10505a/98-3 |
|
Beriate® 2000 |
Zul.-Nr.: |
10505a/98-4 |
9. DATUM DER ERTEILUNG DER ZULASSUNG/VERLÄNGERUNG DER ZULASSUNG
Erstzulassung: 29.01.1998
Letzte Verlängerung der Zulassung: 04.07.2007
10. STAND DER INFORMATION
Datum der letzten Textüberarbeitung: April 2016
11. HERKUNFTSLÄNDER DES BLUTPLASMAS
Belgien, Dänemark, Deutschland, Österreich, Polen, Ungarn, USA
12. VERSCHREIBUNGSSTATUS
Verschreibungspflichtig
CSL Behring Seite 14 von 14