Betafact 500 I.e.
Gebrauchsinformation: Information für Anwender BETAFACT 500 I.E. Pulver und Lösungsmittel zur Herstellung einer Injektionslösung
Blutgerinnungsfaktor IX vom Menschen
Lesen Sie die gesamte Packungsbeilage sorgfältig durch, bevor Sie mit der Anwendung dieses Arzneimittels beginnen, denn sie enthält wichtige Informationen.
- Heben Sie die Packungsbeilage auf. Vielleicht möchten Sie diese später nochmals lesen.
- Wenn Sie weitere Fragen haben, wenden Sie sich an Ihren Arzt oder Apotheker.
- Dieses Arzneimittel wurde Ihnen persönlich verschrieben. Geben Sie es nicht an Dritte weiter. Es kann anderen Menschen schaden, auch wenn diese die gleichen Beschwerden haben wie Sie.
- Wenn Sie Nebenwirkungen bemerken, wenden Sie sich an Ihren Arzt oder Apotheker. Dies gilt auch für Nebenwirkungen, die nicht in dieser Packungsbeilage angegeben sind. Siehe Abschnitt 4.
Was in dieser Packungsbeilage steht
1. Was ist BETAFACT 500 I.E. und wofür wird es angewendet?
2. Was sollten Sie vor der Anwendung von BETAFACT 500 I.E. beachten?
3. Wie ist BETAFACT 500 I.E. anzuwenden?
4. Welche Nebenwirkungen sind möglich?
5. Wie ist BETAFACT 500 I.E. aufzubewahren?
6. Inhalt der Packung und weitere Informationen
1. Was ist BETAFACT 500 I.E. und wofür wird es angewendet?
BETAFACT 500 I.E. ist ein Arzneimittel, das zur Klasse der blutungsstillenden Substanzen gehört. Der Wirkstoff ist humaner Gerinnungsfaktor IX, ein Protein, das auch natürlicherweise im Körper vorkommt. Die Funktion dieses Proteins liegt darin, eine normale Blutgerinnung sicherzustellen und zu verhindern, dass eine Blutung zu lange andauert.
BETAFACT 500 I.E. wird eingesetzt, um einen Mangel des Gerinnungsfaktors IX auszugleichen und somit Blutungen (Hämorrhagien) bei Patienten mit Hämophilie B zu verhindern und zu behandeln.
Hämophilie B ist eine Erbkrankheit, die sich durch Mangel eines Proteins auszeichnet, dem Gerinnungsfaktor IX. Dieser Mangel führt zu Gerinnungsstörungen.
2. Was sollten Sie vor der Anwendung von BETAFACT 500 I.E. beachten?
BETAFACT 500 I.E. darf nicht angewendet werden,
■ wenn Sie allergisch gegen den Faktor IX oder einen der in Abschnitt 6. genannten sonstigen Bestandteile dieses Arzneimittels sind.
■ wenn Sie allergisch gegen Heparin bzw. Heparinderivate sind.
■ bei aktuellem oder aus der Vorgeschichte bekanntem allergisch bedingtem Abfall der Zahl der Blutplättchen (Thrombozytopenie Typ II) aufgrund von Heparin.
■ wenn Ihr Arzt Sie darüber informiert hat, dass Sie allergisch gegen Heparin sind, konsultieren Sie ihn vor Gebrauch dieses Arzneimittels.
Warnhinweise und Vorsichtsmaßnahmen
Bitte sprechen Sie mit Ihrem Arzt oder Apotheker, bevor Sie BETAFACT 500 I.E. anwenden.
Ihr Arzt sollte die möglichen Vorteile der Behandlung mit BETAFACT 500 I.E. abwägen:
Wegen des Risikos einer anormalen Blutgerinnselbildung (thromboembolische Komplikationen)
■ bei Patienten mit Anzeichen von Blutgerinnselabbau (Fibrinolyse)
■ bei Patienten mit multipler Gerinnselbildung im zirkulierenden Blut (disseminierte intravaskuläre Koagulation)
■ bei Neugeborenen
■ bei frischen chirurgischen Eingriffen
■ wenn Ihre Blutgerinnungswerte ungewöhnlich hoch sind
■ wenn eine Lebererkrankung vorliegt
Ihr Arzt wird Sie bitten, eine Blutuntersuchung durchführen zu lassen, um diese Komplikationen möglichst früh zu entdecken.
Risiko allergischer Reaktionen
Angesichts des Allergierisikos (siehe Abschnitt 4: Welche Nebenwirkungen sind möglich) unter der Behandlung mit Faktor IX müssen die ersten Injektionen von BETAFACT 500 I.E. unter ärztlicher Aufsicht erfolgen, um gegebenenfalls eine unmittelbare Allergiebehandlung zu ermöglichen.
Ihr Arzt wird Sie über die Warnzeichen einer allergischen Reaktion informieren (siehe Abschnitt 4: Welche Nebenwirkungen sind möglich). Wenn eine dieser Reaktionen auftritt, muss die Behandlung sofort abgebrochen und der Arzt hinzugezogen werden, um die geeignete Behandlung in Abhängigkeit von Typ und Schwere der Reaktion einzuleiten.
Nach wiederholten Behandlungen mit BETAFACT 500 I.E. kann Ihr Immunsystem auf Faktor IX mit Bildung von Hemmkörpern reagieren (Anti-Faktor-IX-Antikörper). Das Vorkommen dieser Hemmkörper kann die Behandlungswirkung mindern. Ihr Arzt sollte regelmäßige Blutuntersuchungen durchführen, um das Auftreten dieser Hemmkörper zu kontrollieren und ihre Konzentration zu messen.
Zwischen dem Auftreten der Faktor-IX-Hemmkörper und dem Eintreten allergischer Reaktionen konnte ein Zusammenhang festgestellt werden. Daher gilt:
■ wenn sich bei Ihnen nach dem Einsatz von Faktor IX allergische Reaktionen zeigen, müssen Hemmkörpertests durchgeführt werden.
■ wenn Faktor-IX-Hemmkörper nachgewiesen werden, besteht ein größeres Risiko für eine ernsthafte allergische Reaktion während der Injektion von Faktor IX.
BETAFACT 500 I.E. enthält neben Faktor IX auch Spuren anderer menschlicher Proteine. Diese Proteine könnten auch eine Rolle beim Auftreten allergischer Reaktionen spielen.
Informationen zu Sicherheitsmaßnahmen aufgrund der Herkunft von BETAFACT 500 I.E.
BETAFACT 500 I.E. wird aus menschlichem Plasma (der flüssige Teil des Blutes) gewonnen.
Wenn Arzneimittel aus menschlichem Blut oder Plasma gewonnen werden, werden bestimmte Maßnahmen ergriffen, um zu verhindern, dass Infektionen auf Patienten übertragen werden. Diese umfassen:
■ sorgfältige Auswahl der Blut- und Plasmaspender, um sicherzustellen, dass Personen ausgeschlossen werden, die Träger von Infektionen sein können;
■ Test jeder einzelnen Spende und Plasmapools auf Anzeichen von Virusinfektionen;
■ Aufnahme von Schritten in die Verarbeitung von Blut oder Plasma, die Viren inaktivieren oder entfernen können.
Trotz dieser Maßnahmen kann die Möglichkeit einer Übertragung einer Infektion nicht völlig ausgeschlossen werden, wenn medizinische Präparate verabreicht werden, die aus menschlichem Blut oder Plasma hergestellt wurden. Dies gilt auch für unbekannte oder entstehende Viren und andere Arten von Infektionen.
Die eingesetzten Maßnahmen gelten als wirksam gegen umhüllte Viren, wie z. B. das humane Immundefizienzvirus (HIV oder AIDS-Virus), das Hepatitis-B-Virus und das Hepatitis-C-Virus.
Die eingesetzten Maßnahmen sind jedoch gegen nicht-umhüllte Viren, wie z. B. das Hepatitis-A-Virus und das Parvovirus B19, möglicherweise von begrenzter Wirksamkeit. Infektionen mit dem Parvovirus B19 können für Schwangere (Infektion des ungeborenen Kindes) und für Menschen mit Immunschwäche oder bestimmten Arten von Anämie (z. B. Sichelzellenanämie oder hämolytische Anämie) gefährlich sein.
Ihr Arzt wird möglicherweise eine Impfung gegen Hepatitis A und B empfehlen, wenn Sie regelmäßig/wiederholt Faktor-IX-Produkte aus Humanplasma erhalten.
Bei jeder Verabreichung von BETAFACT 500 I.E. wird nachdrücklich empfohlen, den Namen und die Chargenbezeichnung des Präparats zu notieren, damit die verwendeten Chargen registriert werden.
Kinder
Die angeführten Warnhinweise und Vorsichtsmaßnahmen gelten gleichermaßen für Erwachsene und Kinder.
Anwendung von BETAFACT 500 I.E. zusammen mit anderen Arzneimitteln
Informieren Sie Ihren Arzt oder Apotheker, wenn Sie andere Arzneimittel anwenden, kürzlich andere Arzneimittel angewendet haben oder beabsichtigen, andere Arzneimittel anzuwenden, auch wenn es sich um nicht verschreibungspflichtige Arzneimittel handelt.
Es wurden keine Wechselwirkungen zwischen BETAFACT 500 I.E. und anderen Arzneimitteln beobachtet.
Schwangerschaft und Stillzeit
Die Verwendung von BETAFACT 500 I.E. wurde bei schwangeren oder stillenden Frauen nicht untersucht.
Wenn Sie schwanger sind oder stillen, sollte Ihr Arzt den Nutzen der Behandlung mit BETAFACT 500 I.E. beurteilen. Der potenzielle Nutzen dieser Behandlung sollte den damit verbundenen Risiken gegenübergestellt werden.
Wenn Sie schwanger sind oder stillen, oder wenn Sie vermuten, schwanger zu sein oder beabsichtigen, schwanger zu werden, fragen Sie vor der Anwendung dieses Arzneimittels Ihren Arzt oder Apotheker um Rat.
Verkehrstüchtigkeit und Fähigkeit zum Bedienen von Maschinen
Nichts weist darauf hin, dass Faktor IX Auswirkungen auf das Fahrverhalten oder die Fähigkeit zum Bedienen von Maschinen hat.
BETAFACT 500 I.E. enthält Natrium
Dieses Arzneimittel enthält etwa 2,6 mg Natrium pro ml des Produkts (13 mg pro 5-ml-Behälter,
26 mg pro 10-ml-Behälter, 52 mg pro 20-ml-Behälter). Wenn Sie eine kochsalzarme Diät einhalten müssen, sollten Sie dies berücksichtigen.
BETAFACT 500 I.E. enthält Heparin
Dieses Arzneimittel kann allergische Reaktionen hervorrufen und die Anzahl der Blutkörperchen vermindern, was zu einer Blutgerinnungsstörung führen kann.
3. Wie ist BETAFACT 500 I.E. anzuwenden?
Die Behandlung wird von einem Arzt überwacht, der Erfahrungen in der Behandlung von Bluterkrankheiten (Hämophilie) besitzt.
Dosis
Ihr Arzt wird Ihnen die geeignete Dosierung von BETAFACT 500 I.E. nennen.
Die geeignete Dosis und Einnahmehäufigkeit hängt von folgenden Faktoren ab:
■ Ihrem Gewicht,
■ Ausprägung Ihrer Bluterkrankheit (Hämophilie),
■ Ort und Ausmaß der Blutung,
■ Ihrem Gesundheitszustand,
■ und, in bestimmten Fällen, von der Operation, der Sie sich unterziehen möchten (z. B. chirurgischer Eingriff, Zahnextraktion).
Ihr Arzt wird Ihnen empfehlen, im Verlauf der Behandlung Ihr Blut untersuchen zu lassen, zur Kontrolle:
■ der Faktor-IX-Werte,
■ des Auftretens von Faktor-IX-Hemmkörpern.
Aufgrund der Untersuchungsergebnisse kann Ihr Arzt entscheiden, ob Dosis und Häufigkeit Ihrer Injektionen angepasst werden müssen.
Die geeignete Dosis wird in Einheiten (I.E.) angegeben.
Häufigkeit der Verabreichung
Ihr Arzt wird entscheiden, wie oft BETAFACT 500 I.E.-Injektionen verabreicht werden müssen.
Ihr Arzt wird die Häufigkeit der Injektionen der Schwere Ihrer Blutung und der Wirksamkeit der Behandlung anpassen.
Die Tabelle im Abschnitt am Ende der Packungsbeilage, die dem medizinischen Personal vorbehalten ist, erläutert die Häufigkeit und Dauer der Behandlung für verschiedene Situationen.
Art und Weise der Verabreichung
Dieses Arzneimittel ist als intravenöse Infusion zu verabreichen.
Wenn Sie weitere Fragen zur Anwendung dieses Arzneimittels haben, wenden Sie sich an Ihren Arzt oder Apotheker.
Wenn Sie eine größere Menge von BETAFACT 500 I.E. angewendet haben, als Sie sollten
Wenden Sie sich umgehend an Ihren Arzt oder Apotheker.
Jedoch wurde noch über keine Überdosierung mit menschlichem Gerinnungsfaktor IX berichtet.
Wenn Sie die Anwendung von BETAFACT 500 I.E. vergessen haben
Wenden Sie nicht die doppelte Menge an, wenn Sie die vorherige Anwendung vergessen haben.
4. Welche Nebenwirkungen sind möglich?
Wie alle Arzneimittel kann auch dieses Arzneimittel Nebenwirkungen haben, die aber nicht bei jedem auftreten müssen.
Risiko allergischer Reaktionen
• Überempfindlichkeit oder allergische Reaktionen können selten auftreten. In einigen Fällen haben sich diese Reaktionen zu einer schweren allergischen Reaktion entwickelt.
• Allergische Reaktionen können gleichzeitig mit der Entwicklung von Faktor-IX-Hemmkörpern auftreten und können die Nierenfunktion beeinträchtigen (siehe auch Abschnitt 2. „Risiko allergischer Reaktionen“).
Die Warnzeichen einer allergischen Reaktion sind:
• Schwellung von Gesicht und Rachen,
• Gefühl von Brennen und Kribbeln an der Injektionsstelle,
• Schüttelfrost,
• Rötung,
• Juckreiz und Ausschlag,
• niedriger Blutdruck
• extreme Ermüdung (Lethargie),
• Übelkeit, Erbrechen,
• Unruhe,
• beschleunigter Herzschlag,
• Engegefühl in der Brust,
• Kribbeln und Prickeln,
• Giemen (ähnlich wie bei Asthma).
Wenn eine dieser Wirkungen auftritt, brechen Sie die Behandlung sofort ab und wenden Sie sich an einen Arzt, um je nach der Art und der Schwere der Reaktion eine entsprechende Behandlung einzuleiten.
Die folgenden Nebenwirkungen wurden mit BETAFACT in klinischen Studien direkt beobachtet und können selten auftreten (können bis zu 1 von 1.000 Injektionen betreffen):
• Überempfindlichkeit und allergische Reaktionen (siehe auch Abschnitte 2 und 4 „Risiko allergischer Reaktionen“),
• Kopfschmerzen
• Juckreiz,
• allergisches Ödem,
• Übelkeit,
• Reaktion auf die Injektion (Unwohlsein, Brustschmerz),
• Reaktion an der Einstichstelle.
Zwei Fälle aktivitätsneutralisierender Antikörper (Hemmkörper) bei einem zuvor unbehandelten Patienten und bei einem zuvor behandelten Patienten wurden in der Zeit nach Zulassung von BETAFACT gemeldet.
Die folgenden Nebenwirkungen wurden nicht in klinischen Studien mit BETAFACT beobachtet, sondern wurden bei Patienten beobachtet, die Arzneimittel aus derselben Gruppe wie BETAFACT angewendeten:
Blutgerinnsel
Blutgerinnsel können bei der Anwendung von Faktor-IX-Präparaten mit geringem Reinheitsgrad auftreten. Sie können:
• die Versorgung mit Blut und Sauerstoff des Herzens blockieren und einen Herzanfall auslösen.
• die Versorgung mit Blut und Sauerstoff der Lunge blockieren und eine schwere Erkrankung auslösen, die Lungenembolie genannt wird.
• Gerinnsel in einer Vene verursachen (Venenthrombose).
• Blutgerinnsel in den Blutgefäßen im ganzen Körper auslösen (disseminierte intravasale Gerinnung).
BETAFACT enthält hochreinen Faktor IX und wird mit dieser Wirkung selten in Verbindung gebracht.
Entwicklung von Hemmkörpern
• Patienten, die Faktor-IX-Präparate erhalten, können Anti-Faktor-IX-Antikörper entwickeln (sogenannte Hemmkörper - siehe Abschnitt 2 „Risiko allergischer Reaktionen“).
• Diese Hemmkörper wurden in klinischen Studien mit BETAFACT an 11 zuvor unbehandelten Patienten nicht beobachtet.
Meldung von Nebenwirkungen
Wenn Sie Nebenwirkungen bemerken, wenden Sie sich an Ihren Arzt Apotheker oder das medizinische Fachpersonal. Dies gilt auch für Nebenwirkungen, die nicht in dieser Packungsbeilage angegeben sind.
Sie können Nebenwirkungen auch direkt dem Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel, Paul-Ehrlich-Institut, Paul-Ehrlich-Straße 51-59, 63225 Langen, Telefon: +49 6103 77 0, Telefax: +49 6103 77 1234, Website: www.pei.de anzeigen. Indem Sie Nebenwirkungen melden, können Sie dazu beitragen, dass mehr Informationen über die Sicherheit dieses Arzneimittels zur Verfügung gestellt werden.
5. Wie ist BETAFACT 500 I.E. aufzubewahren?
Bewahren Sie dieses Arzneimittel für Kinder unzugänglich auf.
Sie dürfen dieses Arzneimittel nach dem auf dem Etikett und dem Umkarton nach „Verwendbar bis“ angegebenen Verfalldatum nicht mehr verwenden.
Im Kühlschrank lagern (2 °C bis 8 °C). Nicht einfrieren.
Die Durchstechflaschen im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Das Produkt sollte unmittelbar nach der Rekonstitution verwendet werden.
Für den ambulanten Einsatz darf dieses Produkt vor dem Öffnen maximal 6 Monate lang außerhalb des Kühlschranks und bei einer Temperatur von maximal 25 °C aufbewahrt werden. Das Datum, an dem das Arzneimittel aus dem Kühlschrank genommen wird, und das neue Verfalldatum müssen auf dem Umkarton notiert werden. Dieses neue Verfalldatum darf niemals über das ursprüngliche Verfalldatum auf dem Umkarton hinausreichen. Wenn das Arzneimittel vor dem neuen Verfalldatum nicht verbraucht wurde, muss es entsorgt werden.
Sie dürfen dieses Arzneimittel nicht verwenden, wenn die Lösung trüb ist oder Ablagerungen zeigt.
Entsorgen Sie Arzneimittel nicht im Abwasser oder Haushaltsabfall. Fragen Sie Ihren Apotheker, wie das Arzneimittel zu entsorgen ist, wenn Sie es nicht mehr verwenden. Sie tragen damit zum Schutz der Umwelt bei.
6. Inhalt der Packung und weitere Informationen Was BETAFACT 500 I.E. enthält
Der Wirkstoff ist: Blutgerinnungsfaktor IX vom Menschen bei einer Konzentration von 50 I.E./ml nach Zubereitung. Nach Rekonstitution mit 5 ml Wasser für Injektionszwecke enthält eine Durchstechflasche 500 I.E./5 ml humanen Gerinnungsfaktor IX.
Die spezifische Aktivität von BETAFACT 500 I.E. ist ungefähr 110 I.E./mg Protein.
Die sonstigen Bestandteile sind:
Natriumchlorid, Heparin-Natrium, Lysinhydrochlorid, Arginin, Natriumcitrat und Wasser für Injektionszwecke (siehe Abschnitt 2 „Was sollten Sie vor der Anwendung von BETAFACT beachten?“).
Wie BETAFACT 500 I.E. aussieht und Inhalt der Packung
BETAFACT 500 I.E. wird als Pulver und Lösungsmittel in Durchstechflaschen aus Glas zur Herstellung einer Injektionslösung, mit Transfersystem und Filternadel geliefert.
Pharmazeutischer Unternehmer
LFB-BIOMEDICAMENTS 3, avenue des Tropiques BP 305 - LES ULIS 91958 COURTABOEUF Cedex FRANKREICH
Hersteller
LFB-BIOMEDICAMENTS 3, avenue des Tropiques BP 305 - LES ULIS 91958 COURTABOEUF Cedex FRANKREICH
Herkunftsland des Plasmas
Zur Herstellung von BETAFACT 500 I.E. wird Plasma aus Deutschland, Österreich und den USA verwendet.
Dieses Arzneimittel ist in den Mitgliedsstaaten des Europäischen Wirtschaftsraumes (EWR) unter den folgenden Bezeichnungen zugelassen:
Deutschland: BETAFACT 500 I.E Frankreich: BETAFACT 50 UI/ml Griechenland: BETAFACT 50 IU/ml Niederlande: BETAFACT 50 I.E/ml Österreich: BETAFACT 50 I.E./ml Polen: BETAFACT 250 IU/500 IU/1000 IU Portugal: BETAFACT 50 UI/ml
Rumänien: BETAFACT 50 UI/ml, pulbere si solvent pentru solutie injectabila
Slowakei: BETAFACT 50 IU/ml
Spanien: BETAFACT 50 IU/ml
Tschechische Republik: BETAFACT 50 IU/ml
Ungarn: BETAFACT 50 NE/ml
Vereinigtes Königreich: BETAFACT 50 IU/ml
Diese Packungsbeilage wurde zuletzt genehmigt im Mai 2014.
Die folgenden Informationen sind für medizinisches Fachpersonal bestimmt:
Die geeignete Dosis wird in Einheiten (I.E.) angegeben und mithilfe folgender Gleichung berechnet:
Anzahl der zu verabreichenden Einheiten = Körpergewicht (kg) x erwünschter Anstieg des Faktor-IX-Werts (%) (I.E./100 ml) x 0,93
Dosierung
Für jede der unten aufgeführten Blutungsepisoden sollte die Faktor-IX-Aktivität im entsprechenden Zeitraum den angegebenen Wert nicht unterschreiten. Diese Tabelle kann dazu dienen, die wirksame Dosis für folgende Blutungsepisoden und chirurgische Eingriffe festzulegen:
|
Intensität der Blutung/ Art des chirurgischen Eingriffs |
Gewünschte Plasmawerte an Faktor IX (%)(I.E./100ml) |
Häufigkeit der Injektionen (Stunden)/ Behandlungsdauer (Tage) |
|
Hämorrhagien: | ||
|
Frühe Hämarthrose, Muskelblutung oder orale Blutung. |
20 bis 40 |
Wiederholen Sie die Injektion alle 24 Stunden, mindestens 1 Tag lang, bis der Schmerz vergeht oder die Blutung gestoppt wird. |
|
Extensive Hämarthrose, Muskelblutung oder Hämatom. |
30 bis 60 |
Wiederholen Sie die Injektion alle 24 Stunden für 3 bis 4 Tage oder länger, bis der gewöhnliche Bewegungsumfang wieder erreicht wird und der Schmerz aufhört. |
|
Lebensgefährliche Hämorrhagie. |
60 bis 100 |
Wiederholen Sie die Injektion alle 8 bis 24 Stunden, bis die Blutung unter Kontrolle ist. |
|
Chirurgischer Eingriff: | ||
|
Kleiner Eingriff einschließlich Zahnextraktion |
30 bis 60 |
Alle 24 Stunden, mindestens 1 Tag lang, bis die Blutung gestoppt wird. |
|
Großer Eingriff |
80 bis 100 (vor und nach dem |
Wiederholen Sie die Injektion alle 8 bis 24 Stunden, bis eine adäquate Wundheilung |
|
chirurgischen Eingriff) |
erfolgt. Setzen Sie die Behandlung über mindestens 7 weitere Tage fort und halten Sie die Faktor-IX-Aktivität zwischen 30 % und 60 % (I.E./100 ml). |
Unter bestimmten Umständen können größere Mengen von BETAFACT 500 I.E. als die Berechneten erforderlich sein, besonders im Falle der Anfangsdosis.
Während der Behandlung ist die Konzentration des Faktors IX zu überwachen, um eine korrekte Überwachung von Dosis und Verabreichungshäufigkeit der Infusionen zu gewährleisten. Insbesondere bei größeren chirurgischen Eingriffen ist eine präzise Überwachung der Substitutionstherapie mittels Koagulationsanalyse (Aktivität des Plasmafaktors IX) unerlässlich. Patienten können unterschiedlich auf Faktor IX reagieren und unterschiedliche Halbwertszeiten und in vivo Recoveries aufweisen.
Für die Langzeitprophylaxe einer schweren Hämophilie B liegt die empfohlene Dosis bei 20 bis 40 I.E. BETAFACT 500 I.E. pro kg Körpergewicht, die alle 3 bis 4 Tage verabreicht wird.
In bestimmten Fällen, besonders bei jungen Patienten, kann es erforderlich sein, die Dosen zu erhöhen oder die Zeit zwischen zwei Injektionen zu verkürzen.
Patienten sind hinsichtlich des Aufbaus von Faktor-IX-Inhibitoren zu überwachen. Werden die erwarteten Aktivitätskonzentrationen für Faktor IX im Plasma nicht erreicht, oder lässt sich die Blutung mit einer angemessenen Dosis nicht kontrollieren, ist eine Analyse auf Vorliegen eines Faktor-IX-Inhibitors vorzunehmen. Wenn der Inhibitor mit einer Konzentration unter 10 BU (Bethesda Units)/ml vorliegt, kann die Verabreichung zusätzlichen Gerinnungsfaktors IX vom Menschen diesen neutralisieren. Bei Patienten mit einem Inhibitortiter über 10 BU oder mit starker anamnestischer Reaktion sollte der Einsatz von aPCC [(activated) Prothrombin Complex Concentrate] oder FVIIa (activated Factor VII) in Erwägung gezogen werden. Diese Therapien sind von Medizinern zu veranlassen, die Erfahrungen im Bereich der Behandlung von HämophiliePatienten haben.
In klinischen Untersuchungen wurden 11 Kinder unter 6 Jahren mit BETAFACT in Dosierungen behandelt, die denen von Erwachsenen entsprechen.
Das Präparat wird mit Wasser für Injektionszwecke wie unten beschrieben rekonstituiert. Beachten Sie die geltenden Vorschriften für aseptisches Arbeiten.


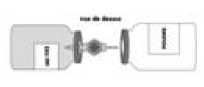

• Falls erforderlich, bringen Sie die beiden Behälter (Pulver und Lösungsmittel) auf Raumtemperatur.
• Nehmen Sie die Schutzkappe vom Lösungsmittelbehälter (Wasser für Injektionszwecke) und vom Pulverbehälter ab.
• Desinfizieren Sie die Oberfläche jedes Stopfens.
• Entfernen Sie die durchsichtige Schutzhülle vom Transfersystem und führen Sie die freigelegte Nadel vollständig durch die Mitte des Stopfens des Lösungsmittelbehälters unter gleichzeitigem Drehen der Nadel ein.
• Entfernen Sie die zweite Schutzhülle vom anderen Ende des Transfersystems.
• Halten Sie beide Behälter horizontal (Entlüftungsdorn zeigt nach oben), drücken Sie schnell das freie Ende der Nadel in die Mitte des Stopfens des Pulverbehälters.
• Vergewissern Sie sich, dass die Nadel immer in das Lösungsmittel eintaucht, um ein vorzeitiges Entweichen des Vakuums zu verhindern.
• Bringen Sie das System unverzüglich in die senkrechte Position, halten Sie dabei den Lösungsmittelbehälter direkt über den Pulverbehälter, um die Übertragung des Lösungsmittels in das Pulver zu ermöglichen.
• Lenken Sie den Strahl des Lösungsmittels während des Transfers auf die gesamte Oberfläche des Pulvers. Versichern Sie sich, dass das Lösungsmittel vollständig überführt wurde.
• Am Ende des Übertragungsvorgangs entweicht das Vakuum automatisch (sterile Luft).
• Entfernen Sie den leeren Behälter (Lösungsmittel) mit dem Transfersystem.
• Schwenken Sie den Behälter unter kreisenden Bewegungen für einige Minuten behutsam, um Schaumbildung zu verhindern, bis das Pulver vollständig gelöst ist.
Das Pulver löst sich im Allgemeinen sofort auf und sollte in weniger als 5 Minuten vollständig gelöst sein.
Die Lösung sollte durchsichtig sein.
Verwenden Sie keine trübe oder Ablagerungen enthaltende Lösung.
Mischen Sie die Lösung nicht mit anderen medizinischen Produkten.
Verdünnen Sie das rekonstituierte Produkt nicht.
Verabreichung:
Ziehen Sie das Produkt unter Verwendung der mitgelieferten Filterkanüle in eine sterile Spritze auf. Entfernen Sie die Filterkanüle von der Spritze.
Setzen Sie eine für intravenöse Injektionen geeignete Kanüle auf die Spritze, drücken die Luft aus der Spritze, desinfizieren die Haut über der Vene und führen die Kanüle ein.
Injizieren Sie intravenös als Einzeldosis höchstens 4 ml/min unmittelbar nach Rekonstitution.
Aufbewahrung nach Rekonstitution:
Aus Gründen der Sterilität sollte das Produkt sofort verwendet werden. Es wurde jedoch eine physiko-chemische Stabilität bis zu 3 Stunden nach der Rekonstitution bei 25 °C gezeigt.