Buprenorphin Acino 35 Mikrogramm/Stunde Transdermales Pflaster
Zul.Nr.: 73818-73820.00.00
Stand: August 2014
Version: 2.0
Buprenorphin-ratiopharm 35 / 52,5 / 70 Mikrogramm/Stunde
T ransdermales Pflaster
Fachinformation
FACHINFORMATION
1. BEZEICHNUNG DES ARZNEIMITTELS
Buprenorphin-ratiopharm 35 Mikrogramm/Stunde Transdermales Pflaster Buprenorphin-ratiopharm 52,5 Mikrogramm/Stunde Transdermales Pflaster Buprenorphin-ratiopharm 70 Mikrogramm/Stunde Transdermales Pflaster
2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
Buprenorphin-ratiopharm 35 Mikrogramm/Stunde Transdermales Pflaster Ein transdermales Pflaster enthält 20 mg Buprenorphin.
Wirkstoffhaltige Fläche: 25 cm2
Nominale Abgaberate: 35 Mikrogramm Buprenorphin pro Stunde Sonstiger Bestandteil mit bekannter Wirkung: Sojaöl (Ph.Eur.) 16 mg
Buprenorphin-ratiopharm 52,5 Mikrogramm/Stunde Transdermales Pflaster Ein transdermales Pflaster enthält 30 mg Buprenorphin.
Wirkstoffhaltige Fläche: 37,5 cm2
Nominale Abgaberate: 52,5 Mikrogramm Buprenorphin pro Stunde Sonstiger Bestandteil mit bekannter Wirkung: Sojaöl (Ph.Eur.) 24 mg
Buprenorphin-ratiopharm 70 Mikrogramm/Stunde Transdermales Pflaster Ein transdermales Pflaster enthält 40 mg Buprenorphin.
Wirkstoffhaltige Fläche: 50 cm2
Nominale Abgaberate: 70 Mikrogramm Buprenorphin pro Stunde Sonstiger Bestandteil mit bekannter Wirkung: Sojaöl (Ph.Eur.) 32 mg
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
3. DARREICHUNGSFORM
Transdermales Pflaster
Buprenorphin-ratiopharm 35 Mikrogramm/Stunde Transdermales Pflaster
Hautfarbenes rechteckiges Pflaster, mit vier abgerundeten Ecken, mit der Aufschrift „Buprenorphin
35 pg/h“ bedruckt.
Buprenorphin-ratiopharm 52,5 Mikrogramm/Stunde Transdermales Pflaster
Hautfarbenes rechteckiges Pflaster, mit vier abgerundeten Ecken, mit der Aufschrift „Buprenorphin
52,5 pg/h“ bedruckt.
Buprenorphin-ratiopharm 70 Mikrogramm/Stunde Transdermales Pflaster
Hautfarbenes rechteckiges Pflaster, mit vier abgerundeten Ecken, mit der Aufschrift „Buprenorphin 70 pg/h“ bedruckt.
Jedes Pflaster ist in einzelverschweißten Beuteln verpackt.
4. KLINISCHE ANGABEN
4.1 Anwendungsgebiete
Mäßig starke bis starke Tumorschmerzen und starke Schmerzen bei ungenügender Wirksamkeit nicht-opioider Schmerzmittel.
Buprenorphin-ratiopharm ist für die Behandlung von akuten Schmerzen nicht geeignet.
4.2 Dosierung und Art der Anwendung Dosierung
Patienten über 18 Jahre
Die Dosierung von Buprenorphin soll grundsätzlich der Situation des einzelnen Patienten (Schmerzstärke, Leidensdruck, individuelle Reaktion) angepasst werden. Es ist jeweils die niedrigste ausreichend schmerzlindernde Dosierung anzustreben. Für eine entsprechend adaptive Behandlung stehen drei Stärken des transdermalen Pflasters zur Verfügung: Buprenorphin-ratiopharm 35 Mikrogramm/Stunde, Buprenorphin-ratiopharm 52,5 Mikrogramm/Stunde und Buprenorphin-ratiopharm 70 Mikrogramm/Stunde.
Wahl der Anfangsdosis
Bei Patienten ohne vorherige Anwendung von Analgetika soll die Behandlung mit der niedrigsten Stärke des transdermalen Pflasters (Buprenorphin 35 Mikrogramm/h) begonnen werden. Patienten die bereits mit einem Analgetikum der WHO-Stufe I (Nicht-Opioid) oder der WHO-Stufe II (schwach wirksames Opioid) behandelt wurden, sollten entsprechend mit Buprenorphin-ratiopharm 35 Mikrogramm/Stunde beginnen. Gemäß den Empfehlungen der WHO kann abhängig von der medizinischen Gesamtsituation des Patienten die Einnahme eines Nicht-Opioid-Analgetikums beibehalten werden.
Bei Umstellung von einem Analgetikum der WHO-Stufe III (stark wirksames Opioid) auf Buprenorphin-ratiopharm empfiehlt es sich zur Minimierung eines Schmerzrückfalls, bei der Wahl der initialen Stärke des transdermalen Pflasters die Vorbehandlung nach Art des Wirkstoffs, Art der Anwendung und der durchschnittlichen Tagesdosierung zu berücksichtigen.
Allgemein ist es empfehlenswert die Dosis individuell zu titrieren, indem mit der kleinsten Pflasterstärke (Buprenorphin-ratiopharm 35 Mikrogramm/Stunde) begonnen wird. Klinische Erfahrungen haben gezeigt, dass Patienten, die zuvor mit höheren Tagesdosen eines stark wirksamen Opioids behandelt wurden (in der Größenordnung von etwa 120 mg oral appliziertem Morphin), die Therapie auch mit der nächst größeren Pflasterstärke beginnen können (s. auch Abschnitt 5.1).
Um die individuelle Dosisfindung innerhalb einer angemessenen Zeit zu ermöglichen, sollten während der Dosistitration geeignete, schnell freisetzende Analgetika zur Verfügung gestellt werden.
Die erforderliche Dosisstärke von Buprenorphin-ratiopharm muss auf die individuellen Bedürfnisse des einzelnen Patienten abgestimmt und regelmäßig überprüft werden.
Da die Buprenorphin-Konzentrationen im Serum, sowohl bei nicht mit Analgetika vorbehandelten als auch bei derart vorbehandelten Patienten, nach Applikation des ersten Buprenorphin-ratiopharm transdermalen Pflasters langsam ansteigen, ist ein rascher Wirkungseintritt unwahrscheinlich. Eine erste Bewertung der schmerzlindernden Wirkung sollte aus diesem Grund frühestens nach den ersten 24 Stunden erfolgen.
Die analgetische Vormedikation (mit Ausnahme von transdermalen Opioiden) sollte nach Umstellung auf Buprenorphin-ratiopharm über die ersten 12 Stunden in unveränderter Dosierung gegeben werden, und bei Bedarf eine geeignete Zusatzmedikation in den folgenden 12 Stunden.
Dosistitrierung und Erhaltungstherapie
Buprenorphin-ratiopharm kann bis zu maximal 72 Stunden angewendet werden. Buprenorphin-ratiopharm sollte spätestens nach 72 Stunden (3 Tagen) ersetzt werden. Die Dosistitrierung sollte individuell durchgeführt werden, bis die analgetische Wirkung erreicht ist. Ist die Analgesie am Ende des ersten Applikationszeitraums unzureichend, kann die Dosis erhöht werden, entweder indem mehr als ein transdermales Pflaster der gleichen Stärke appliziert wird oder indem zur nächst höheren Pflasterstärke übergegangen wird. Unabhängig von der Pflasterstärke sollten gleichzeitig nicht mehr als zwei transdermale Pflaster angewendet werden.
Vor Applikation der nächst höheren Pflasterstärke von Buprenorphin-ratiopharm sollte die Gesamtmenge an Opioiden, die gegebenenfalls zusätzlich zu dem bisherigen transdermalen Pflaster verabreicht wurde, bedacht werden. D. h. die Gesamtmenge an benötigten Opioiden muss bedacht und die Dosierung muss entsprechend angepasst werden. Patienten, die während der Erhaltungstherapie einer zusätzlichen Analgetikagabe bedürfen (z. B. bei Durchbruchschmerzen), können alle 24 Stunden 1-2 Sublingualtabletten mit jeweils 0,2 mg Buprenorphin zusätzlich zu dem transdermalen Pflaster einnehmen. Bei regelmäßiger Notwendigkeit von zusätzlich 0,4 mg bis 0,6 mg Buprenorphin sublingual sollte die nächst höhere Pflasterstärke eingesetzt werden.
Patienten unter 18 Jahre
Da Buprenorphin-ratiopharm bei Patienten, die jünger als 18 Jahre sind, bisher nicht untersucht wurde, wird eine Anwendung in dieser Altersgruppe nicht empfohlen.
Ältere Patienten
Bei älteren Patienten ist keine Änderung der Dosierung von Buprenorphin-ratiopharm erforderlich. Patienten mit Niereninsuffizienz
Da sich die Pharmakokinetik von Buprenorphin bei Nierenversagen nicht verändert, ist die Anwendung bei Niereninsuffizienz, einschließlich Dialysepatienten, möglich.
Patienten mit Leberinsuffizienz
Buprenorphin wird in der Leber metabolisiert. Die Intensität und Dauer seiner Wirkung kann bei Patienten mit Leberfunktionsstörungen verändert sein. Daher sollten solche Patienten bei Behandlung mit Buprenorphin-ratiopharm sorgfältig überwacht werden.
Art der Anwendung
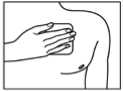
Buprenorphin-ratiopharm soll auf nicht gereizte, gereinigte, unbehaarte, flache Hautpartien und nicht auf Hautstellen mit größeren Narben aufgebracht werden. Vorzugsweise erfolgt die Applikation am Oberkörper, auf der oberen Rückenpartie bzw. unterhalb des Schlüsselbeins auf der Brust. Eventuell vorhandene Haare sollen nicht rasiert, sondern mit einer Schere entfernt werden. Falls die Applikationsstelle gereinigt werden muss, sollte dies mit Wasser geschehen. Dabei dürfen weder Seife noch andere Reinigungsmittel benutzt werden. Auf die für das Aufkleben des transdermalen Pflasters ausgewählte Hautstelle sollen keine Dermatika aufgetragen werden, die das Kleben von Buprenorphin-ratiopharm beeinträchtigen könnten.
Die Haut muss vor der Applikation vollkommen trocken sein. Buprenorphin-ratiopharm soll unmittelbar nach dem Herausnehmen aus dem Beutel appliziert werden.
Buprenorphin-ratiopharm soll kontinuierlich bis zu 3 Tage getragen werden. Nachdem das vorangegangene transdermale Pflaster entfernt wurde, ist ein neues Buprenorphin-ratiopharm
Buprenorphin-ratiopharm 35 / 52,5 / 70 Mikrogramm/Stunde
Zul.Nr.: 73818-73820.00.00 Stand: August 2014 Version: 2.0
T ransdermales Pflaster
Fachinformation
Pflaster an einer anderen Stelle aufzukleben. Bevor auf dieselbe Hautstelle ein neues transdermales Pflaster appliziert wird, sollte mindestens 1 Woche vergangen sein.
1. Der Beutel darf erst unmittelbar vor der Anwendung geöffnet werden.
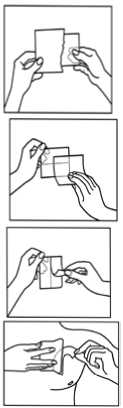
2. Zu Beginn wird die lose Trennfolie entfernt.
3. Danach wird eine Hälfte der Abziehfolie des Pflasters entfernt ohne dabei die Klebeschicht zu berühren.
4. Die andere Hälfte der Abziehfolie wird nach dem Aufkleben des transdermalen Pflasters auf die ausgewählte Hautstelle entfernt.
5. Das transdermale Pflaster muss für ungefähr 30 - 60 Sekunden mit der flachen Hand auf die Haut gepresst werden um sicher zu sein, dass das gesamte transdermale Pflaster auf der Haut klebt, besonders an den Rändern.
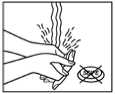
6. Nach der Anwendung des Pflasters sollen die Hände ohne Reinigungsmittel gewaschen werden.
Vorausgesetzt, das transdermale Pflaster wurde richtig angebracht, ist ein Ablösen sehr unwahrscheinlich. Duschen, baden oder schwimmen während des Tragens ist möglich, allerdings sollte jegliche Hitzeeinwirkung (z. B. Sauna, Infrarot-Bestrahlung, elektrische Wärmdecken, Wärmflaschen) vermieden werden.
Sollte sich das transdermale Pflaster vor dem nächsten Wechsel lösen, darf dasselbe transdermale Pflaster nicht nochmals aufgeklebt werden. Es ist ein neues Pflaster zu applizieren.
Wechsel des transdermalen Pflasters
- Das alte transdermale Pflaster wird vorsichtig abgenommen.
- Die klebrigen Enden des Pflasters werden gegeneinander gefaltet mit, wobei die Klebefläche innen liegen sollte.
- Das Pflaster muss mit Bedacht entsorgt werden.
- Ein neues transdermales Pflaster wird auf eine andere geeignete Hautstelle aufgeklebt (wie oben beschrieben). Dieselbe Hautstelle kann erst nach zwei weiteren Anwendungen wieder beklebt werden.
Dauer der Anwendung
Buprenorphin-ratiopharm sollte auf keinen Fall länger als therapeutisch notwendig angewendet werden. Wenn entsprechend Art und Schwere der Erkrankung eine länger dauernde Schmerzbehandlung mit Buprenorphin-ratiopharm erforderlich erscheint, sollte sorgfältig und in kurzen Abständen regelmäßig überprüft werden (gegebenenfalls durch Einlegen von Anwendungspausen), ob und inwieweit ein medizinisches Erfordernis weiter besteht.
Absetzen von Buprenorphin-ratiopharm
Nach Entfernen von Buprenorphin-ratiopharm fällt die Buprenorphin-Konzentration im Serum kontinuierlich ab, wodurch die schmerzlindernde Wirkung noch über einen bestimmten Zeitraum erhalten bleibt. Dies muss bedacht werden, wenn nach Buprenorphin-ratiopharm ein anderes Opioid
angewendet werden soll. Allgemein gilt, dass ein nachfolgendes Opioid nicht innerhalb der nächsten 24 Stunden nach Absetzen von Buprenorphin-ratiopharm angewendet werden darf. Derzeit liegen nur sehr wenige Informationen über die Initialdosis eines anderen Opioids nach Absetzen von Buprenorphin-ratiopharm vor.
4.3 Gegenanzeigen
Buprenorphin-ratiopharm darf nicht angewendet werden:
- bei Überempfindlichkeit gegen den Wirkstoff Buprenorphin, Soja, Erdnuss oder einen der sonstigen Bestandteile (zu den sonstigen Bestandteilen siehe Abschnitt 6.1)
- bei opioidabhängigen Patienten und zur Behandlung bei Drogensubstitution
- bei Krankheitszuständen, bei denen eine schwere Störung des Atemzentrums und der Atemfunktion vorliegt oder sich entwickeln kann
- bei Patienten, die MAO-Hemmer erhalten oder innerhalb der letzten 2 Wochen erhalten haben (siehe Abschnitt 4.5)
- bei Patienten mit Myasthenia gravis
- bei Patienten mit Delirium tremens
4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Buprenorphin-ratiopharm darf nur mit besonderer Vorsicht angewendet werden:
- bei Patienten mit akuter Alkoholintoxikation, zerebralen Anfallsleiden, Kopfverletzung, bei Schock, bei Bewusstseinsstörungen unbekannter Genese
- bei Zuständen mit erhöhtem Hirndruck ohne Möglichkeit der Beatmung
- bei Patienten mit eingeschränkter Atemfunktion, da Buprenorphin gelegentlich eine Atemdepression verursachen kann
- bei Patienten mit eingeschränkter Atemfunktion bzw. unter gleichzeitiger Behandlung mit Arzneimitteln, die auch eine Atemdepression auslösen können
Buprenorphin hat ein wesentlich niedrigeres Abhängigkeitspotential als reine Opioid-Agonisten. In Studien mit Buprenorphin-ratiopharm an gesunden Probanden und Patienten wurden keine Entzugserscheinungen beobachtet. Nach einer Langzeitanwendung von Buprenorphin-ratiopharm können jedoch Entzugssymptome, die einem Opiatentzug ähnlich sind, nicht völlig ausgeschlossen werden (siehe Abschnitt 4.8). Diese Symptomatik umfasst: Erregung, Angst, Nervosität, Schlaflosigkeit, Hyperkinesie, Zittern und gastrointestinale Beschwerden.
Bei Patienten, die Opioide missbräuchlich anwenden, kann die Substitution mit Buprenorphin Entzugserscheinungen verhindern. Dies hat gelegentlich zu einem Buprenorphin-Missbrauch geführt. Bei Patienten mit Neigung zu Arzneimittel-/Drogenmissbrauch ist deshalb entsprechende Vorsicht geboten.
Buprenorphin wird in der Leber metabolisiert. Die Intensität und Dauer seiner Wirkung kann bei Patienten mit Leberfunktionsstörungen verändert sein. Daher sollten solche Patienten bei Behandlung mit Buprenorphin-ratiopharm einer sorgfältigen Kontrolle unterliegen.
Buprenorphin ist im Wettbewerb von der Welt Anti-Doping Behörde als Substanz verboten worden.
Patienten mit Fieber/äußere Wärmeeinwirkung
Fieber und äußere Wärmeeinwirkung können zu einer erhöhten Permeabilität der Haut führen. Bei Anwendung von Buprenorphin-ratiopharm können unter diesen Bedingungen theoretisch die Buprenorphin-Konzentrationen im Serum erhöht sein. Bei Patienten mit Fieber, oder anderweitig bedingter erhöhter Hauttemperatur, sollte deshalb bei Behandlung mit Buprenorphin-ratiopharm auf verstärkte Opioidreaktionen geachtet werden.
4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Bei Gabe von MAO-Hemmstoffen innerhalb der letzten 14 Tage vor einer Gabe des Opioids Pethidin sind lebensbedrohliche Wechselwirkungen beobachtet worden, die das Zentralnervensystem sowie Atmungs- und Kreislauffunktion betrafen. Dieselben Wechselwirkungen mit MAO-Hemmstoffen sind bei Buprenorphin-ratiopharm nicht auszuschließen (siehe Abschnitt 4.3).
Bei gemeinsamer Anwendung von Buprenorphin-ratiopharm mit anderen Opioiden, Anästhetika, Hypnotika, Sedativa, Antidepressiva, Neuroleptika und generell mit Arzneimitteln, die dämpfende Wirkungen auf Atmung und das zentrale Nervensystem haben, kann es zur gegenseitigen Verstärkung von ZNS-Effekten kommen. Dies gilt auch für die Anwendung mit Alkohol.
Bei gemeinsamer Anwendung mit CYP 3A4 Inhibitoren oder Induktoren kann die Effektivität von Buprenorphin-ratiopharm gesteigert (Inhibitoren z.B. Ketoconazol) oder gemindert (Induktoren z.B. Phenobarbital, Carbamazepin, Phenytoin und Rifampicin) werden.
4.6 Fertilität, Schwangerschaft und Stillzeit
Schwangerschaft
Es liegen nur wenige Daten für die Anwendung von Buprenorphin-ratiopharm bei Schwangeren vor. Tierexperimentelle Studien haben eine Reproduktionstoxizität gezeigt (siehe Abschnitt 5.3). Das potentielle Risiko für den Menschen ist nicht bekannt.
Gegen Ende der Schwangerschaft können hohe Dosen von Buprenorphin auch nach kurzer Anwendungsdauer Atemdepression bei Neugeborenen induzieren.
Die Anwendung von Buprenorphin über längere Zeit während der letzten drei Schwangerschaftsmonate kann bei Neugeborenen ein Entzugssyndrom hervorrufen.
Daher sollte Buprenorphin-ratiopharm während der Schwangerschaft und bei Frauen im gebärfähigen Alter, die keine zuverlässige Methode zur Verhütung praktizieren, nicht angewendet werden.
Stillzeit
Buprenorphin wird beim Menschen in die Muttermilch ausgeschieden.
Studien an Ratten haben gezeigt, dass Buprenorphin die Laktation hemmen kann.
Buprenorphin-ratiopharm sollte während der Stillzeit nicht angewendet werden.
4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Buprenorphin-ratiopharm kann auch bei bestimmungsgemäßen Gebrauch das Reaktionsvermögen so weit verändern, dass die Fähigkeit zur aktiven Teilnahme am Straßenverkehr oder zum Bedienen von Maschinen beeinträchtigt wird. Dies gilt insbesondere im Zusammenwirken mit anderen zentral wirksamen Mitteln, einschließlich Alkohol, Beruhigungsmitteln, Sedativa und Hypnotika.
Patienten, die Buprenorphin-ratiopharm transdermales Pflaster anwenden, sollten währenddessen und bis zu 24 Stunden nach der Entfernung des transdermalen Pflasters kein Fahrzeug fahren oder Maschinen bedienen.
4.8 Nebenwirkungen
Die folgenden Nebenwirkungen wurden nach der Anwendung von Buprenorphin in klinischen Studien und im Rahmen der Produktüberwachung berichtet.
Bei der Bewertung von Nebenwirkungen werden folgende Häufigkeiten zugrunde gelegt:
Sehr häufig (> 1/10)
Häufig (> 1/100 bis < 1/10)
Gelegentlich (> 1/1.000 bis < 1/100)
Selten (> 1/10.000 bis < 1/1.000)
Sehr selten (< 1/10.000)
nicht bekannt (auf Grundlage der verfügbaren Daten nicht abschätzbar)
Die am häufigsten berichteten systemischen Nebenwirkungen waren Übelkeit und Erbrechen. Von lokalen Nebenwirkungen wurden am häufigsten Erytheme und Juckreiz gemeldet.
Erkrankungen des Immunsystems
Sehr selten: schwere allergische Reaktionen
Stoffwechsel- und Ernährungsstörungen Selten: Appetitverlust
Psychiatrische Erkrankungen
Gelegentlich: Verwirrtheit, Schlafstörungen, Unruhe
Selten: Psychotomimetische Effekte (z. B. Halluzinationen, Angstzustände, Alpträume),
Libidoverminderung
Sehr selten: Abhängigkeit, Stimmungsschwankungen
Erkrankungen des Nervensystems
Häufig:
Gelegentlich:
Selten:
Sehr selten:
Schwindel, Kopfschmerzen Sedierung, Somnolenz
Konzentrationsstörungen, Sprachstörung, Benommenheit, Gleichgewichtsstörungen, Parästhesien (z. B. Hautprickeln und brennende Hautirritationen)
Faszikuläre Muskelzuckungen, Geschmacksstörungen
Augenerkrankungen
Selten: Sehstörungen, verschwommenes Sehen, Augenlidödeme
Sehr selten: Miosis
Erkrankungen des Ohrs und des Labyrinths Sehr selten: Ohrenschmerzen
Herz- und Gefäßerkrankungen
Gelegentlich: Kreislaufstörungen (wie Hypotonie oder in seltenen Fällen Kreislaufkollaps) Selten: Hitzegefühl
Erkrankungen der Atemwege, des Brustraums und Mediastinums Häufig: Dyspnoe
Selten: Atemdepression
Sehr selten: Hyperventilation, Schluckauf
Erkrankungen des Gastrointestinaltrakts Sehr häufig: Übelkeit Häufig: Erbrechen, Verstopfung
Gelegentlich: Mundtrockenheit Selten: Sodbrennen
Sehr selten: Brechreiz
Erkrankungen der Haut und des Unterhautzellgewebes
Sehr häufig: Erytheme, Juckreiz
Häufig: Exantheme, Schwitzen
Gelegentlich: Ausschlag
Selten: Urtikaria
Sehr selten: Pusteln, Bläschen
Erkrankungen der Nieren und Harnwege Gelegentlich: Harnverhaltung, Miktionsstörungen
Erkrankungen der Geschlechtsorgane und der Brustdrüse Selten: Erektionsschwäche
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort Häufig: Ödeme, Müdigkeit
Gelegentlich: Abgeschlagenheit
Selten: Entzugserscheinungen, Reaktionen am Verabreichungsort
Sehr selten: Brustschmerz
In einigen Fällen traten verzögert allergische Reaktionen mit deutlichen Anzeichen einer Entzündung auf. In diesen Fällen sollte die Behandlung mit Buprenorphin-ratiopharm beendet werden.
Buprenorphin hat ein geringes Abhängigkeitsrisiko. Nach Absetzen von Buprenorphin-ratiopharm sind Entzugssymptome unwahrscheinlich, da Buprenorphin sehr langsam von den Opiatrezeptoren dissoziiert, und die Buprenorphin-Konzentrationen im Serum kontinuierlich abnehmen (gewöhnlich über einen Zeitraum von 30 Stunden nach Entfernen des letzten transdermalen Pflasters). Nach Langzeitanwendung von Buprenorphin-ratiopharm können Entzugssymptome wie bei Opiatentzug jedoch nicht gänzlich ausgeschlossen werden. Zur Symptomatik gehören Unruhe, Angst, Nervosität, Schlaflosigkeit, Hyperkinesie, Zittern und Magen-Darmstörungen.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Arzneimittel und Medizinprodukte, Abt. Pharmakovigilanz, Kurt-Georg-Kiesinger-Allee 3, D-53175 Bonn, Website: www.bfarm.de anzuzeigen.
4.9 Überdosierung
Buprenorphin besitzt eine große therapeutische Breite. Da Buprenorphin kontrolliert in kleinen Mengen in den Blutkreislauf abgegeben wird, ist es unwahrscheinlich, dass hohe bzw. toxische Buprenorphin-Konzentrationen im Blut auftreten. Die maximale Buprenorphin-Konzentration im Serum nach Applikation von Buprenorphin-ratiopharm 70 Mikrogramm/Stunde Transdermales Pflaster ist um das 6-fache niedriger als nach intravenöser Applikation der therapeutischen Dosis von 0,3 mg Buprenorphin.
Symptome
Grundsätzlich treten nach einer Buprenorphin-Überdosis ähnliche Symptome auf, wie sie auch bei anderen zentralwirksamen Analgetika (Opioide) zu erwarten sind. Sie umfassen Atemdepression, Sedierung, Somnolenz, Übelkeit, Erbrechen, Kreislaufkollaps und ausgeprägte Miosis.
Behandlung
Es sind die allgemeinen Notfallmaßnahmen anzuwenden. Die Atemwege sind freizuhalten (Aspiration!). Atmung und Kreislauf sind abhängig von den Symptomen aufrecht zu erhalten. Die Möglichkeiten, die durch Buprenorphin hervorgerufene Atemdepression durch Naloxon aufzuheben, sind begrenzt. Hierzu ist Naloxon in hohen Dosen als wiederholter Bolus oder als Infusion zu verabreichen (z. B. mit einem Bolus [intravenös] zu Beginn von 1-2 mg. Nach Erreichen eines adäquaten antagonistischen Effekts ist die Anwendung einer Infusion nötig, um die Plasmaspiegel von Naloxon konstant zu halten). Eine ausreichende Ventilation muss daher sichergestellt werden.
5. PHARMAKOLOGISCHE EIGENSCHAFEN
5.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Opioide, Oripavin-Derivate.
ATC-Code: N02AE01
Buprenorphin ist ein stark wirksames Opioid mit agonistischer Aktivität am p-Opioidrezeptor und antagonistischer Aktivität am kappa-Opioidrezeptor. Die Eigenschaften von Buprenorphin scheinen vergleichbar mit denen von Morphin, die Substanz weist jedoch spezifische pharmakologische und klinische Besonderheiten auf.
Darüber hinaus muss der Einfluss zahlreicher Faktoren wie z. B. Indikation, klinische Situation, Applikationsweg und interindividuelle Variabilität auf die analgetische Wirksamkeit bei einem Vergleich verschiedener Analgetika berücksichtigt werden.
In der täglichen klinischen Praxis werden unterschiedliche Opioide mittels einer relativen Potenz eingeordnet, obwohl dies eine starke Vereinfachung darstellt.
Die relative Potenz von Buprenorphin in unterschiedlichen Darreichungsformen und verschiedenen klinischen Situationen ist in der Literatur wie folgt beschrieben worden:
• Morphin peroral: Buprenorphin intramuskulär als 1 : 67 bis 150 (Einmalgabe; Akutschmerzmodell)
• Morphin peroral: Buprenorphin sublingual als 1 : 60 bis 100 (Einmalgabe, Akutschmerzmodell, Mehrfachgabe, chronischer Schmerz, Tumorschmerz)
• Morphin peroral: Buprenorphin transdermal als 1 : 75 bis 115 (Mehrfachgabe, chronischer Schmerz)
Die Nebenwirkungen sind denen anderer starker Opioid-Analgetika vergleichbar. Das Abhängigkeitspotential von Buprenorphin scheint niedriger als das von Morphin.
5.2 Pharmakokinetische Eigenschaften
Allgemeine Eigenschaften des Wirkstoffs
Die Bindung von Buprenorphin an Plasmaproteine beträgt etwa 96 %.
Buprenorphin wird in der Leber zu N-Dealkylbuprenorphin (Norbuprenorphin) und glukuronidierten Metaboliten verstoffwechselt. 2/3 des Wirkstoffs werden unverändert mit den Faezes ausgeschieden und 1/3 als Konjugate von unverändertem oder dealkyliertem Buprenorphin über die Harnwege. Es gibt Hinweise auf eine enterohepatische Rezirkulation.
Untersuchungen an trächtigen und nicht-trächtigen Ratten haben gezeigt, dass Buprenorphin sowohl die Blut-Hirnschranke als auch die Plazentaschranke passiert. Nach parenteraler Gabe waren die Konzentrationen im Gehirn (nur unverändertes Buprenorphin vorhanden) 2-3-fach höher als nach oraler Gabe.
Nach intramuskulärer oder oraler Verabreichung kumulierte Buprenorphin offenbar im Gastrointestinallumen des Fötus - vermutlich aufgrund der biliären Ausscheidung, da der enterohepatische Kreislauf nicht entwickelt ist.
Eigenschaften von Buprenorphin-ratiopharm bei gesunden Probanden
Nach Applikation von Buprenorphin-ratiopharm wird Buprenorphin über die Haut aufgenommen. Die kontinuierliche Abgabe von Buprenorphin in den Kreislauf erfolgt durch kontrollierte Freisetzung aus dem anhaftenden Polymer-Matrixsystem.
Nach der ersten Applikation von Buprenorphin-ratiopharm steigt die Buprenorphin-Konzentration im Plasma langsam an und erreicht die minimal-effektive Konzentration von 100 pg/ml nach 4 bis 12 Stunden. In Studien an Probanden mit Buprenorphin-ratiopharm 35 Mikrogramm/Stunde wurde eine durchschnittliche maximale Konzentration Cmax von 273 pg/ml und eine durchschnittliche tmax von 34 h ermittelt. Bei Studien mit Buprenorphin-ratiopharm 70 Mikrogramm/Stunde wurde eine durchschnittliche maximale Konzentration Cmax von 425 pg/ml und eine durchschnittliche tmax von 29 h ermittelt.
Nach Entfernen von Buprenorphin-ratiopharm fielen die Buprenorphin-Konzentrationen im Plasma kontinuierlich mit einer Halbwertszeit von etwa 25 h (im Mittel 24-27 Stunden) ab. Die kontinuierliche Resorption von Buprenorphin aus dem Hautdepot führt zu einer langsameren Elimination als nach intravenöser Gabe.
5.3 Präklinische Daten zur Sicherheit
Die Standarduntersuchungen zur Toxikologie ergaben keine Hinweise auf ein besonderes Gefahrenpotential für den Menschen. In Studien mit wiederholter Gabe von Buprenorphin bei Ratten wurde eine reduzierte Körpergewichtszunahme beobachtet.
Studien zur Fertilität und allgemeinen Reproduktionsfähigkeit an Ratten zeigten keine nachteiligen Effekte. Untersuchungen an Ratten und Kaninchen ergaben Hinweise auf Fetotoxizität und einen erhöhten Postimplantationsverlust.
Studien an Ratten haben ein vermindertes intrauterines Wachstum, Entwicklungsverzögerungen einiger neurologischer Funktionen und eine hohe peri-postnatale Sterblichkeit der Neugeborenen nach Behandlung der Muttertiere während der Trächtigkeit bzw. der Laktation ergeben.
Es liegen Hinweise vor, dass Geburtsschwierigkeiten und eine reduzierte Milchproduktion zu diesen Effekten beigetragen haben. Anzeichen für Embryotoxizität einschließlich Teratogenität gab es weder bei Ratten noch bei Kaninchen.
Untersuchungen in-vitro und in-vivo zum mutagenen Potenzial von Buprenorphin zeigten keine klinisch relevanten Effekte.
Langzeituntersuchungen an Ratte und Maus ergaben keine für den Menschen relevanten Hinweise auf ein karzinogenes Potenzial.
Die vorhandenen toxikologischen Daten wiesen nicht auf ein allergenes Potenzial der Hilfsstoffe der transdermalen Pflaster hin.
6. PHARMAZEUTISCHE ANGABEN
6.1 Liste der sonstigen Bestandteile
Aloe-vera Blätter, FE mit raffiniertem Sojabohnenöl und all-rac-a-Tocopherolacetat (Ph.Eur.)
Wirkstoffhaltige adhäsive Matrix: Styrol-Butadien-Styrol (SBS) und Poly(butadien-block-styrol)(11,3:17) mit Klebstoff und Antioxidans
Trägerschicht: Pigmentiertes Polyethylen, thermoplastisches Harz und aluminiumbedampftes überzogenes Polyester, blaue Beschriftungstinte
Schutzfolie mit Abziehhilfe: Polyesterfilm, einseitig silikonisiert (wird vor dem Aufkleben abgezogen)
6.2 Inkompatibilitäten
Nicht zutreffend
6.3 Dauer der Haltbarkeit
18 Monate
6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Nicht über 25 °C lagern.
Nicht einfrieren.
6.5 Art und Inhalt des Behältnisses
Jedes transdermale Pflaster ist mit einer losen silikonisierten PETP Folie bedeckt und einzeln in einem Beutel versiegelt. Der Beutel besteht aus PETP, Aluminiumfolie und Polyethylen.
Die Packungen enthalten 4, 5, 8, 10, 16 oder 20 einzeln versiegelte transdermale Pflaster. Klinikpackung: 24 einzeln versiegelte transdermale Pflaster.
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung
Größere Mengen an Buprenorphin verbleiben auch nach der Benutzung im transdermalen Pflaster. Daher müssen benutzte transdermale Pflaster mit der Klebefläche nach innen zusammengefaltet und entsorgt oder wieder in die Apotheke zurück gebracht werden. Jedes nicht benutzte Arzneimittel sollte weggeworfen oder zurück in die Apotheke gebracht werden.
7. INHABER DER ZULASSUNG
Acino AG Am Windfeld 35 83714 Miesbach
8. ZULASSUNGSNUMMERN
Buprenorphin-ratiopharm 35 Mikrogramm/Stunde Transdermales Pflaster
73818.00. 00
Buprenorphin-ratiopharm 52,5 Mikrogramm/Stunde Transdermales Pflaster
73819.00. 00
Buprenorphin-ratiopharm 70 Mikrogramm/ Stunde Transdermales Pflaster
73820.00. 00
9. DATUM DER ERTEILUNG DER ZULASSUNG/VERLÄNGERUNG DER ZULASSUNG
Datum der Erteilung der Zulassung: 23. April 2010
Datum der letzten Verlängerung der Zulassung: 28. Februar 2014
10. STAND DER INFORMATION
August 2014
11. VERKAUFSABGRENZUNG
Verschreibungspflichtig, Betäubungsmittel