Calcihexal 100
Fachinformation
1. BEZEICHNUNG DES ARZNEIMITTELS
CalciHEXAL® 100 I.E./ml Injektionslösung
Wirkstoff: Calcitonin (Lachs)
2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
1 Ampulle mit 1 ml Injektionslösung enthält 100 I.E. Calcitonin (Lachs).
Dabei entspricht 1 I.E. 0,167 Mikrogramm des Wirkstoffs.
Sonstiger Bestandteil mit bekannter Wirkung:
Natriumverbindungen
Vollständige Auflistung der sonstigen Bestandteile siehe Abschnitt 6.1.
3. DARREICHUNGSFORM
Injektionslösung
Klare, farblose, wässrige Flüssigkeit
4. KLINISCHE ANGABEN
4.1 Anwendungsgebiete
• Prävention eines akuten Verlustes an Knochenmasse nach einer plötzlichen Immobilisation, zum Beispiel bei Patienten mit einer vor kurzem festgestellten osteoporotischen Fraktur.
• Für die Behandlung des Morbus Paget nur bei Patienten, die auf Behandlungsalternativen nicht ansprechen oder für die solche Behandlungsmöglichkeiten nicht geeignet sind, zum Beispiel Patienten mit schwerer Nierenfunktionsstörung.
• Behandlung von Hyperkalzämie infolge von malignen Erkrankungen.
4.2 Dosierung und Art der Anwendung
Zur subkutanen oder intramuskulären Injektion oder zur intravenösen Infusion bei Personen, die mindestens 18 Jahre alt sind.
Um die Häufigkeit von Übelkeit und Erbrechen herabzusetzen, die vor allem zu Beginn der Behandlung auftreten können, kann Calcitonin vor dem Schlafengehen angewendet werden.
Aufgrund der Nachweise für ein erhöhtes Risiko für maligne Erkrankungen bei der Langzeitanwendung von Calcitonin (siehe Abschnitt 4.4) sollte die Behandlungsdauer bei allen Anwendungsgebieten auf die kürzest mögliche Zeit beschränkt werden und es sollte die kleinste wirksame Dosis angewendet werden.
Prävention eines akuten Verlustes an Knochenmasse nach einer plötzlichen Immobilisation, zum Beispiel bei Patienten mit einer vor kurzem festgestellten osteoporotischen Fraktur Die empfohlene Dosis beträgt 100 I.E. pro Tag oder 50 I.E. zweimal täglich subkutan oder intramuskulär. Vor der erneuten Mobilisation kann die Dosis auf 50 I.E. täglich reduziert werden. Die empfohlene Behandlungsdauer beträgt 2 Wochen und sollte aufgrund des
Zusammenhangs zwischen einem erhöhten Krebsrisiko und der Langzeitanwendung von Calcitonin 4 Wochen keinesfalls überschreiten.
Morbus Paget
Die empfohlene Dosierung beträgt 100 I.E. pro Tag subkutan oder intramuskulär. Klinische und biochemische Besserungen wurden jedoch auch mit einem MinimaldosisBehandlungsschema von 50 I.E. dreimal wöchentlich erzielt. Die Dosierung muss den individuellen Bedürfnissen des Patienten angepasst werden. Die Behandlung sollte abgebrochen werden, sobald der Patient angesprochen hat und die Symptome beseitigt sind. Die Behandlungsdauer sollte aufgrund der Nachweise für ein erhöhtes Risiko für maligne Erkrankungen bei der Langzeitanwendung von Calcitonin im Normalfall 3 Monate nicht überschreiten. In Ausnahmefällen, z. B. bei Patienten mit drohenden Spontanfrakturen, kann die Behandlungsdauer bis auf ein empfohlenes Maximum von 6 Monaten ausgedehnt werden.
Regelmäßige erneute Behandlungen können bei diesen Patienten erwogen werden. Bei diesen erneuten Behandlungen sollten der potenzielle Nutzen und die Nachweise für ein erhöhtes Risiko für maligne Erkrankungen bei der Langzeitanwendung von Calcitonin (siehe Abschnitt 4.4) berücksichtigt werden.
Die Wirkung von Calcitonin kann anhand der Messung geeigneter Marker des Knochenumbaus, wie beispielsweise der alkalischen Phosphatase im Serum oder der Hydroxyprolin- bzw. Desoxypyridinolin-Werte im Harn, überwacht werden.
Hyperkalzämie infolge von Malignität
Die empfohlene Initialdosis beträgt 100 I.E. alle 6-8 Stunden mittels subkutaner oder intramuskulärer Injektion. Außerdem kann Calcitonin nach vorhergehender Rehydratation intravenös injiziert werden. Sollte das Ansprechen hierauf nach ein oder zwei T agen nicht zufriedenstellend sein, kann die Dosis auf maximal 400 I.E. alle 6-8 Stunden erhöht werden.
In schweren Fällen oder bei Notfällen können über eine Zeitspanne von mindestens 6 Stunden bis zu 10 I.E./kg Körpergewicht in 500 ml 0,9%iger (w/v) Natriumchlorid-Lösung als intravenöse Infusion verabreicht werden. Die Dosierung von CalciHEXAL sollte den individuellen Bedürfnissen des Patienten angepasst werden.
Ältere Menschen
Erfahrungen mit der Anwendung von Calcitonin bei älteren Patienten ergeben keine Hinweise auf eine schlechtere Verträglichkeit oder auf einen geänderten Dosierungsbedarf.
Patienten mit Leberfunktionsstörungen
Erfahrungen mit der Anwendung von Calcitonin bei Patienten mit Leberfunktionsstörungen ergeben keine Hinweise auf eine schlechtere Verträglichkeit oder auf einen geänderten Dosierungsbedarf.
Patienten mit Nierenfunktionsstörungen
Die metabolische Clearance ist bei Patienten mit terminalem Nierenversagen sehr viel geringer als bei gesunden Probanden. Die klinische Relevanz dieses Befundes ist jedoch nicht bekannt (siehe Abschnitt 5.2).
Kinder und Jugendliche
Es liegen keine ausreichenden Daten vor, um eine Anwendung von Calcitonin bei Kindern mit Erkrankungen, die auf eine Osteoporose im Kindesalter zurückzuführen sind, zu befürworten. Die Anwendung von Calcitonin wird daher bei Kindern und Jugendlichen bis zu 18 Jahren nicht empfohlen.
4.3 Gegenanzeigen
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Calcitonin ist bei Patienten mit Hypokalzämie kontraindiziert.
4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Da Calcitonin ein Peptid ist, besteht die Möglichkeit systemischer allergischer Reaktionen. Allergische Reaktionen, einschließlich Einzelfälle von anaphylaktischem Schock, wurden bei Patienten beschrieben, die Calcitonin erhalten haben. Solche Reaktionen sollten von generalisierten oder lokalen Rötungen (Flush) unterschieden werden. Hierbei handelt es sich um häufige, nicht-allergische Wirkungen von Calcitonin (siehe Abschnitt 4.8).
Bei Patienten mit Verdacht auf eine Überempfindlichkeit gegen Calcitonin sollte vor Beginn der Behandlung ein Hauttest durchgeführt werden.
Auswertungen randomisierter kontrollierter Studien an Patienten mit Osteoarthritis und Osteoporose haben gezeigt, dass Calcitonin mit einem statistisch signifikanten Anstieg des Krebsrisikos im Vergleich zu mit Placebo behandelten Patienten verbunden ist. Die Erhöhung des absoluten Krebsrisikos bei Patienten, die mit Calcitonin im Vergleich zu Placebo behandelt wurden, variierte für die Dauertherapie in diesen Studien zwischen 0,7 und 2,4 %. Die Patienten in diesen Studien wurden mit oralen oder intranasalen Darreichungsformen behandelt. Es ist jedoch wahrscheinlich, dass ein erhöhtes Risiko auch für die subkutane, intramuskuläre oder intravenöse Verabreichung von Calcitonin besteht. Dies ist besonders bei Langzeitanwendung der Fall, da die systemische Exposition mit Calcitonin für diese Patienten höher sein dürfte als bei anderen Darreichungsformen.
CalciHEXAL enthält Natrium, aber weniger als 1 mmol (23 mg) Natrium pro 1 ml Injektionslösung.
4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Die Serum-Calciumspiegel können zu Behandlungsbeginn vorübergehend unter die Normalwerte absinken. Dies trifft insbesondere für Patienten mit ungewöhnlich hohen Knochenumsatzraten zu. Dieser Effekt wird mit der Verringerung der Osteoklastenaktivität abgeschwächt. Bei Patienten unter gleichzeitiger Behandlung mit Herzglykosiden oder Calcium-Kanalblockern ist jedoch Vorsicht geboten. Die Dosierungen dieser Arzneimittel müssen möglicherweise angesichts der Tatsache, dass ihre Wirkungen durch Veränderungen der zellulären Elektrolytkonzentrationen modifiziert werden können, erneut eingestellt werden.
Bei gleichzeitiger Anwendung von Calcitonin und Bisphosphonaten können additive, Calciumsenkende Effekte auftreten.
Die gleichzeitige Anwendung von Calcitonin und Lithium kann zu einer Verringerung des Lithium-Plasmaspiegels führen. Die Dosierung von Lithium muss möglicherweise angepasst werden.
Sonstige Wechselwirkungen
Da Calcitonin ein Peptid ist, kann es zu Adsorption an Kunststoffen und Infusionsbestecken kommen. Dies birgt die Möglichkeit, dass der Patient nur eine reduzierte Gesamtdosis erhält. Das häufige Überprüfen des klinischen Ansprechens und der Laborwerte, einschließlich der Messung des Serum-Calciumspiegels, wird empfohlen, insbesondere in frühen Phasen der Behandlung.
4.6 Fertilität, Schwangerschaft und Stillzeit
Schwangerschaft
Zu Calcitonin wurden keine Studien an Schwangeren durchgeführt. Calcitonin darf während der Schwangerschaft nur dann angewendet werden, wenn die Behandlung vom Arzt für absolut unerlässlich gehalten wird.
Stillzeit
Es liegen keine Informationen darüber vor, ob Calcitonin beim Menschen in die Muttermilch übertritt. Bei Tieren wurde gezeigt, dass Calcitonin die Milchbildung hemmt und in die Muttermilch übergeht (siehe Abschnitt 5.3). Deshalb wird empfohlen, während der Behandlung nicht zu stillen.
Fertilität
Es liegen keine Daten über einen potenziellen Einfluss von Calcitonin auf die Fertilität beim Menschen vor.
4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Es liegen keine Studien über die Auswirkungen von Calcitonin auf die Fahrtüchtigkeit und auf das Bedienen von Maschinen vor. Calcitonin kann Müdigkeit, Schwindel und Sehstörungen hervorrufen (siehe Abschnitt 4.8). Dadurch kann die Reaktionsfähigkeit der Patienten beeinträchtigt werden. Die Patienten müssen daher auf die Möglichkeit des Auftretens dieser Effekte hingewiesen werden. In diesem Fall sollten die Patienten weder Auto fahren noch Maschinen bedienen.
4.8 Nebenwirkungen
Die am häufigsten beobachteten Nebenwirkungen sind Übelkeit, Erbrechen und Hautrötung. Sie sind dosisabhängig und treten häufiger nach i.v.- als nach i.m.- oder s.c.- Verabreichung auf.
Nebenwirkungen, die aus verschiedenen Quellen, einschließlich aus klinischen Studien und aus Erfahrungsberichten nach Markteinführung stammen, sind gemäß MedDRA-Systemorganklassen aufgelistet. Innerhalb jeder Systemorganklasse sind die Nebenwirkungen nach abnehmender Häufigkeit ihres Auftretens sortiert. Innerhalb jeder Häufigkeitsgruppe werden die Nebenwirkungen nach abnehmendem Schweregrad angegeben.
Bei den Häufigkeitsangaben zu Nebenwirkungen werden folgende Kategorien zugrunde gelegt:
Sehr häufig (> 1/10)
Häufig (> 1/100 bis < 1/10)
Gelegentlich (> 1/1.000 bis < 1/100)
Selten (> 1/10.000 bis < 1/1.000)
Sehr selten (< 1/10.000)
Nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar)
Gutartige, bösartige und unspezifische Neubildungen (einschl. Zysten und Polypen) Häufig:
• bösartige Tumoren (bei Langzeitanwendung)
Erkrankungen des Immunsystems Gelegentlich:
• Überempfindlichkeitsreaktionen Sehr selten:
• schwere allergische Reaktionen wie beispielsweise Bronchospasmus
• Anschwellen von Zunge und Rachenraum
• anaphylaktischer Schock
Stoffwechsel- und Ernährungsstörungen Selten:
• vorübergehende Senkung des Serum-Calciums 1 Nicht bekannt:
• Hypokalzämie
Erkrankungen des Nervensystems Häufig:
• Schwindel
• Kopfschmerzen
• Störungen des Geschmacksempfindens Nicht bekannt:
• Tremor
Augenerkrankungen
Gelegentlich:
• Sehstörungen
Gefäßerkrankungen Sehr häufig:
• Rötung (im Gesicht oder am Oberkörper) 2 Gelegentlich:
• Bluthochdruck
Erkrankungen des Gastrointestinaltrakts Sehr häufig:
• Übelkeit mit oder ohne Erbrechen 3 Häufig:
• Durchfall
• Bauchschmerz
Erkrankungen der Haut und des Unterhautzellgewebes Gelegentlich:
• generalisierter Hautausschlag
• Juckreiz Nicht bekannt:
• Urtikaria
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen Häufig:
• Skelettmuskelschmerzen einschließlich Arthralgien
Erkrankungen der Nieren und Harnwege Gelegentlich:
• übermäßige Harnausscheidung
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort Häufig:
• Müdigkeit Gelegentlich:
• grippeähnliche Erkrankung
• Ödeme (im Gesicht, an den Extremitäten und generalisiert)
• Reaktionen am Ort der Injektion
Untersuchungen
Selten:
• Bildung neutralisierender Antikörper gegen Calcitonin 4
Die Häufigkeit der oben genannten Nebenwirkungen basiert teilweise auf den Ergebnissen von klinischen Studien mit Calcitonin Nasenspray.
1 Bei Patienten mit einem hohen Knochenstoffwechsel (Morbus Paget oder junge Patienten) kann 4-6 Stunden nach der Anwendung eine vorübergehende, üblicherweise asymptomatische Senkung des Serum-Calciums auftreten.
2 Hautrötung (im Gesicht oder am Oberkörper) ist keine allergische Reaktion, sondern auf die pharmakologische Wirkung zurückzuführen und tritt für gewöhnlich innerhalb von 1020 Minuten nach der Anwendung auf.
3 Übelkeit mit oder ohne Erbrechen tritt bei ca. 10 % der mit Calcitonin behandelten Patienten auf. Dieser Effekt ist verstärkt zu Beginn der Behandlung zu beobachten und tendiert dazu, sich bei fortgesetzter Anwendung oder bei Dosisreduktion abzumildern oder ganz zu verschwinden. Falls erforderlich, kann ein Antiemetikum gegeben werden. Übelkeit/Erbrechen sind seltener, wenn die Injektionen am Abend oder nach einer Mahlzeit gegeben werden.
4 Bildung von neutralisierenden Antikörpern gegen Calcitonin: Das Entstehen dieser Antikörper steht normalerweise nicht mit einem Verlust der klinischen Wirksamkeit in Zusammenhang, auch wenn ihr Vorhandensein bei einem kleinen Prozentsatz der Patienten nach der Langzeitbehandlung mit Calcitonin zu einem herabgesetzten Ansprechen auf das Präparat führen könnte. Das Vorhandensein von Antikörpern scheint nicht mit den nur selten auftretenden allergischen Reaktionen in Beziehung zu stehen. Eine Down-Regulation von Calcitonin-Rezeptoren kann ebenfalls ein Grund für eine verminderte klinische Ansprechbarkeit bei einem kleinen Prozentsatz von Patienten während der Langzeitbehandlung sein.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-RisikoVerhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem
Bundesinstitut für Arzneimittel und Medizinprodukte Abt. Pharmakovigilanz Kurt-Georg-Kiesinger-Allee 3 D-53175 Bonn Website: www.bfarm.de
anzuzeigen.
4.9 Überdosierung
Es ist bekannt, dass Übelkeit, Erbrechen, Hautrötung (Flush) und Schwindel dosisabhängig auftreten, wenn Calcitonin parenteral gegeben wird. In einzelnen Dosen wurden bis zu 10.000 I.E. Calcitonin zur Injektion gegeben, ohne dass andere Nebenwirkungen als Übelkeit und Erbrechen oder eine übersteigerte pharmakologische Wirkung aufgetreten wären.
Wenn Anzeichen einer Überdosierung auftreten, sollte eine symptomatische Behandlung erfolgen.
5.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Nebenschilddrüsen-Antagonisten ATC-Code: H05B A01 Calcitonin (Lachs, synthetisch)
Die pharmakologischen Eigenschaften der synthetischen und der rekombinanten Proteine sind erwiesenermaßen qualitativ und quantitativ äquivalent.
Als calcitropes Hormon hemmt Calcitonin die Knochenresorption aufgrund einer direkten Wirkung auf die Osteoklasten. Durch die Hemmung der Osteoklastenaktivität über spezifische Rezeptoren verringert Calcitonin die Knochenresorption. In pharmakologischen Studien konnte eine analgetische Wirkung von Calcitonin am Tiermodell nachgewiesen werden.
Bei Erkrankungen mit erhöhter Knochenresorption wie Morbus Paget oder bei einem akuten Verlust an Knochenmasse nach plötzlicher Immobilisation reduziert Calcitonin deutlich die Knochenumbaurate.
Histomorphologische Studien sowohl am Knochen des Menschen wie auch des Tieres haben nachgewiesen, dass die Anwendung von Calcitonin nicht zu Mineralisationsdefekten führt.
Sowohl bei gesunden Probanden als auch bei Patienten mit Erkrankungen des Skelettsystems einschließlich Morbus Paget und Osteoporose wurden nach der Behandlung mit Calcitonin Verringerungen der Knochenresorption festgestellt, was auf der Grundlage einer Herabsetzung der Hydroxyprolin- und Desoxypyridinolin-Werte im Harn ermittelt wurde.
Die eine Senkung des Calciums hervorrufende Wirkung des Calcitonins basiert sowohl auf einem verringerten Efflux von Calcium aus dem Knochen in die Extrazellularflüssigkeit als auch auf der Hemmung der Calcium-Rückresorption durch die Nierentubuli.
Gleichzeitig hemmt Calcitonin die renale Phosphat-Rückresorption und steigert die Aktivierungsrate des Vitamin D.
5.2 Pharmakokinetische Eigenschaften
Resorption
Calcitonin wird schnell resorbiert (Resorptionshalbwertszeit 10-15 min).
Plasmaspitzenspiegel werden innerhalb der ersten Stunde nach der Gabe erreicht. Nach subkutaner Verabreichung werden die Plasmaspitzenspiegel nach ca. 23 Minuten erreicht. Beim Menschen ist die Bioverfügbarkeit nach der subkutanen und intramuskulären Injektion hoch und für beide Anwendungen ähnlich (71 % bzw. 66 %).
Verteilung
Nach parenteraler Anwendung von 100 I.E. Calcitonin liegt der Plasmaspitzenspiegel zwischen ca. 200 und 400 pg/ml. Die Plasmaprotein-Bindung beträgt 30-40 %.
Biotransformation
Tierstudien haben gezeigt, dass Calcitonin nach parenteraler Anwendung primär durch Proteolyse in der Niere verstoffwechselt wird. Die Metaboliten besitzen nicht die spezifische biologische Aktivität von Calcitonin.
Elimination
Calcitonin besitzt eine kurze Eliminationshalbwertzeit von ca. 1 Stunde nach der intramuskulären und 1-1,5 Stunden nach der subkutanen Verabreichung. Calcitonin wird nahezu ausschließlich in den Nieren abgebaut, wobei pharmakologisch inaktive Fragmente des Moleküls entstehen. Deshalb ist die metabolische Clearance bei Patienten mit terminalem Nierenversagen sehr viel geringer als bei gesunden Freiwilligen. Die klinische Relevanz dieses Befundes ist jedoch nicht bekannt.
Linearität
Es besteht eine Beziehung zwischen der subkutan injizierten Dosis Calcitonin und den Plasmaspitzenspiegeln. Höhere Blutspiegel können mit einer erhöhten Inzidenz von Übelkeit und Erbrechen einhergehen.
5.3 Präklinische Daten zur Sicherheit
Es wurden die üblichen präklinischen Untersuchungen zur chronischen Toxizität, Reproduktionstoxizität, Mutagenität und Kanzerogenität an Labortieren durchgeführt.
Calcitonin besitzt weder ein embryotoxisches, teratogenes noch mutagenes Potenzial.
Bei Ratten, die synthetisches Calcitonin über 1 Jahr erhielten, wurde über eine erhöhte Inzidenz von Hypophysentumoren berichtet. Man nimmt an, dass dieser Effekt Speziesspezifisch ist und keine klinische Relevanz besitzt.
Es ist nicht bekannt, ob Calcitonin die Plazentaschranke passiert.
Bei mit Calcitonin behandelten, laktierenden Tieren wurde eine Unterdrückung der Milchbildung beobachtet. Calcitonin tritt in die Milch über.
6. PHARMAZEUTISCHE ANGABEN
6.1 Liste der sonstigen Bestandteile
• Essigsäure 99 %
• Natriumacetat-Trihydrat
• Natriumchlorid
• Wasser für Injektionszwecke
6.2 Inkompatibilitäten
Inkompatibilitäten sind bisher nicht bekannt.
6.3 Dauer der Haltbarkeit
1 Jahr
Haltbarkeit nach Verdünnung
Die chemische und physikalische Stabilität der gebrauchsfertigen Zubereitung wurde für 24 Stunden bei Raumtemperatur sowie für 72 Stunden bei 2-8 °C nachgewiesen. Aus mikrobiologischer Sicht sollte die gebrauchsfertige Zubereitung sofort verwendet werden. Wenn die gebrauchsfertige Zubereitung nicht sofort verwendet wird, ist der Anwender für die Dauer und die Bedingungen der Aufbewahrung verantwortlich. Sofern die Herstellung der gebrauchsfertigen Zubereitung nicht unter kontrollierten und validierten aseptischen Bedingungen erfolgt, ist diese nicht länger als 24 Stunden bei 2-8 °C aufzubewahren.
Haltbarkeit nach Anbruch
Nicht verwendete Reste sind zu verwerfen.
6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Die Ampullen im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Im Kühlschrank (2-8 °C) lagern.
Aus mikrobiologischer Sicht sollte das Arzneimittel sofort, nachdem es Raumtemperatur angenommen hat, zur Injektion verwendet werden. Zur Infusion sollte es unmittelbar nach der Verdünnung in 0,9%iger (w/v) Natriumchloridlösung bzw. 5%iger Glucose-Lösung in Weichkunststoff-PVC-Beuteln verwendet werden.
Weitere Hinweise siehe Abschnitte 6.3 und 6.6.
6.5 Art und Inhalt des Behältnisses
Klarglasampullen (Typ I Glas, Ph.Eur.) mit je 1 ml Injektionslösung
Packungen mit 5, 10, 20 und 50 Ampullen
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung
CalciHEXAL soll optisch begutachtet werden. Falls die Lösung nicht klar und farblos ist, Partikel enthält oder beschädigt ist, ist das Arzneimittel nicht anzuwenden.
Die Verdünnung mit Natriumchlorid-Lösung 0,9 % oder Glucose-Lösung 5 % sollte immer erst unmittelbar vor der Anwendung in Weichkunststoff-PVC-Beuteln zubereitet werden.
Die Ampullen sind nur zur einmaligen Anwendung. Verbleibender Inhalt muss entsorgt werden. Für eine intramuskuläre oder subkutane Anwendung sollte das Arzneimittel vor der Anwendung Raumtemperatur angenommen haben.
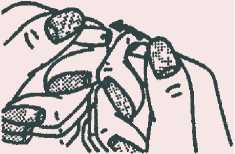
Hinweis zur Handhabung der OPC (one-point-cut)-Ampullen Anfeilen nicht mehr erforderlich.
Punkt nach oben. Im Ampullenspieß befindliche Lösung durch Klopfen oder Schütteln nach unten fließen lassen.
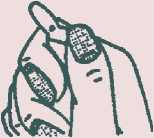
Ampullenspieß vom Punkt nach hinten wegbrechen.
7. INHABER DER ZULASSUNG
Hexal AG Industriestraße 25 83607 Holzkirchen Telefon: (08024) 908-0 Telefax: (08024) 908-1290 E-Mail: medwiss@hexal.com
8. ZULASSUNGSNUMMER
27952.01.00
Datum der Erteilung der Zulassung 6. Februar 1996
Datum der letzten Verlängerung der Zulassung 27. September 2006
10. STAND DER INFORMATION
März 2016
11. VERKAUFSABGRENZUNG
Verschreibungspflichtig