Cil 160 Mg
CiL® 160 mg
FACHINFORMATION
1. BEZEICHNUNG DES ARZNEIMITTELS
CiL 160 mg Hartkapseln
2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
1 Hartkapsel enthält 160 mg Fenofibrat.
Vollständige Auflistung der sonstigen Bestandteile siehe Abschnitt 6.1.
3. DARREICHUNGSFORM
Hartkapsel.
4. KLINISCHE ANGABEN
4.1 Anwendungsgebiete
CiL 160 mg ist angezeigt als unterstützende Behandlung neben einer Diät oder anderen nicht medikamentösen Therapien (z. B. sportlicher Betätigung, Gewichtsabnahme) für folgende Erkrankungen:
- schwere Hypertriglyceridämie mit oder ohne niedrige HDL-Cholesterinwerte,
- gemischte Hyperlipidämie, wenn ein Statin kontraindiziert ist oder nicht vertragen wird,
- bei gemischter Hyperlipidämie bei Patienten mit hohem kardiovaskulärem Risiko zusätzlich zu einem Statin, wenn Triglycerid- und HDL-Cholesterinwerte nicht ausreichend kontrolliert werden können.
4.2 Dosierung und Art der Anwendung
Während der Therapie mit diesem Arzneimittel sollte die Diät fortgesetzt werden. Das Ansprechen auf die Behandlung sollte durch Bestimmung der Serumlipidwerte überwacht werden.
Wird nach mehrmonatiger Behandlung mit Fenofibrat (z. B. 3 Monate) keine ausreichende lipidsenkende Wirkung erreicht, sind ergänzende oder andere therapeutische Maßnahmen in Betracht zu ziehen.
Dosierung
Erwachsene
Empfohlene Tagesdosis: 1 Hartkapsel CiL 160 mg (entsprechend 160 mg Fenofibrat) täglich.
Patienten, die mit einer Einzeldosis von 200 mg mikronisiertem Fibrat behandelt werden, können ohne weitere Dosisanpassung auf 1 Hartkapsel zu 160 mg umgestellt werden.
Besondere Patientengruppen
Ältere Patienten (ab 65 Jahren)
Eine Dosisanpassung ist nicht notwendig. Die Einnahme der üblichen Dosis wird empfohlen, außer bei eingeschränkter Nierenfunktion mit einer geschätzten (estimated) glomerulären Filtrationsrate (eGFR) < 60 ml/min/1,73 m2 (siehe „Patienten mit eingeschränkter Nierenfunktion“).
Patienten mit eingeschränkter Nierenfunktion
Bei stark eingeschränkter Nierenfunktion mit einer eGFR < 30 ml/min/1,73 m2 darf Fenofibrat nicht eingenommen werden.
Bei einer eGFR zwischen 30 und 59 ml/min/1,73 m2 sollte die tägliche Dosis 100 mg Fenofibrat (Standard) oder 67 mg mikronisiert nicht überschreiten.
Wenn in Nachuntersuchungen die eGFR dauerhaft unter 30 ml/min/1,73 m2 fällt, muss die Einnahme von Fenofibrat abgebrochen werden.
Patienten mit eingeschränkter Leberfunktion
Aufgrund von fehlenden Daten ist die Anwendung von CiL 160 mg bei Patienten mit eingeschränkter Leberfunktion nicht zu empfehlen.
Kinder und Jugendliche
Die Unbedenklichkeit und Wirksamkeit von Fenofibrat bei Kindern und Jugendlichen unter 18 Jahren ist nicht hinreichend nachgewiesen. Es liegen keine Studien vor. Aus diesem Grund wird die Anwendung von Fenofibrat bei Kindern und Jugendlichen unter 18 Jahren nicht empfohlen.
Art der Anwendung
Die Hartkapsel soll unzerkaut zu einer der Mahlzeiten eingenommen werden.
4.3 Gegenanzeigen
- Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile,
- Leberinsuffizienz (einschließlich biliärer Zirrhose und unerklärbar persistierender Leberfunktionsabnormität wie z. B. persistierende Erhöhung der Serumtransaminasen),
- bekannte Gallenblasenerkrankungen,
- schwere Niereninsuffizienz (geschätzte glomeruläre Filtrationsrate < 30 ml/min/1,73 m2),
- chronische oder akute Pankreatitis mit Ausnahme einer akuten Pankreatitis aufgrund schwerer Hypertriglyceridämie,
- bekannte photoallergische oder phototoxische Reaktionen unter der Behandlung mit Fibraten oder Ketoprofen.
4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Sekundäre Ursachen einer Hyperlipidämie
Vor der Erwägung einer Therapie mit Fenofibrat sollten sekundäre Ursachen einer Hyperlipidämie wie schlecht eingestellter Diabetes mellitus, Hypothyreose, nephrotisches Syndrom, Dysproteinämien, obstruktive Lebererkrankung oder Alkoholismus adäquat therapiert werden. Sekundäre Ursachen einer Hyperlipidämie können bei einer pharmakologischen Therapie mit Diuretika, Betablockern, Estrogenen, Gestagenen, kombinierten oralen Kontrazeptiva, Immunsuppressiva und Proteaseinhibitoren auftreten. In diesen Fällen sollte geprüft werden, ob es sich um eine primäre oder sekundäre Hyperlipidämie handelt (möglicher Anstieg der Lipidwerte durch diese Arzneimittel).
Leber
Wie bei anderen lipidsenkenden Arzneimitteln wurde unter der Therapie mit Fenofibrat bei einigen Patienten über einen Anstieg der Transaminasen berichtet. In der Mehrzahl der beobachteten Fälle war der Anstieg vorübergehend, geringfügig und asymptomatisch. Es wird empfohlen, die Transaminasenspiegel während des ersten Behandlungsjahres in 3-monatigen Intervallen und später in regelmäßigen Abständen zu überprüfen.
Patienten, bei denen ein erhöhter Transaminasenspiegel festgestellt wird, sollten sorgfältig überwacht werden. Steigen die Spiegel von ASAT (SGOT) und ALAT (SGPT) auf mehr als das Dreifache des oberen Normwertes an, ist die Behandlung abzubrechen. Bei Auftreten von Symptomen, die auf eine Hepatitis hinweisen (z. B. Ikterus, Juckreiz), sowie bestätigte labordiagnostische Untersuchungen, ist Fenofibrat abzusetzen.
Bauchspeicheldrüse
Unter der Behandlung mit Fenofibrat wurde über das Auftreten einer Pankreatitis berichtet (siehe Abschnitte 4.3 und 4.8). Bei Patienten mit einer schweren Hypertriglyceridämie kann dies auf eine nicht ausreichende Wirksamkeit des Arzneimittels, eine direkte Arzneimittelwirkung oder auf einen Sekundäreffekt zurückzuführen sein, der über eine Cholelithiasis mit Verschluss des Ductus choledochus vermittelt wird.
Muskulatur
Bei der Anwendung von Fibraten und anderen Lipidsenkern wurde über Myotoxizität und in seltenen Fällen über Rhabdomyolyse - mit oder ohne Nierenversagen - berichtet. Die Inzidenz dieser Erkrankung steigt im Falle einer Hypalbuminämie und einer vorausgegangenen Niereninsuffizienz.
Ein erhöhtes Risiko, an Rhabdomyolyse zu erkranken, besteht bei Patienten mit prädisponierenden Faktoren für Myopathie und/oder Rhabdomyolyse: Patienten in einem Alter von mehr als 70 Jahren, Muskelerkrankungen in der Vorgeschichte oder Familienanamnese, Nierenfunktionsstörungen, Hypothyreose und bei hohem Alkoholkonsum. Für diesen Patientenkreis ist eine sorgfältige Überwachung des Nutzen-Risiko-Verhältnisses einer Fenofibrat-Therapie erforderlich.
Diffuse Myalgien, Myositis, Muskelkrämpfe, Muskelschwäche und/oder ein erheblicher Anstieg der Kreatinphosphokinase (CPK) (Anstieg über das Fünffache des oberen Normwertes) deuten auf eine Myotoxizität hin. Das Arzneimittel ist in diesen Fällen abzusetzen.
Das Risiko einer Myotoxizität kann sich erhöhen, wenn dieses Arzneimittel zusammen mit einem anderen Fibrat oder einem HMG-CoA-Reduktase-Hemmer (Statin) kombiniert wird. Dies gilt insbesondere, wenn bereits Muskelerkrankungen bestehen. Daher sollte die Kombination von Fenofibrat mit einem HMG-CoA-Reduktase-Hemmer oder einem anderen Fibrat auf Patienten mit schwerer kombinierter Hyperlipidämie und hohem kardiovaskulärem Risiko, bei denen bislang noch keine Muskelerkrankungen aufgetreten sind, beschränkt werden, und diese Patienten sollten streng auf eine mögliche Myotoxizität hin überwacht werden.
Nierenfunktion
CiL 160 mg ist kontraindiziert bei stark eingeschränkter Nierenfunktion (siehe Abschnitt 4.3).
CiL 160 mg sollte bei Patienten mit leichter bis mäßiger Niereninsuffizienz mit Vorsicht angewendet werden. Eine Dosisanpassung ist erforderlich bei Patienten mit einer eGFR zwischen 30 und 59 ml/min/1,73 m2 (siehe Abschnitt 4.2).
Reversible Kreatininwerterhöhungen im Blut wurden bei Patienten, die eine Fenofibrat-Monotherapie oder eine Kombination mit Statinen erhalten haben, beobachtet. Die Kreatininwerterhöhung war im Allgemeinen über die Zeit stabil, Anzeichen eines weiteren Anstiegs wurden bei einer Langzeittherapie nicht beobachtet. Nach Beendigung der Behandlung wurde ein Rückgang auf die Ausgangswerte beobachtet.
In klinischen Studien hatten 10 % der Patienten bei der Kombinationsbehandlung von Fenofibrat und Simvastatin einen auf die Ausgangswerte bezogenen Kreatininanstieg um mehr als 30 pmol/l im Vergleich zu 4,4 % der Patienten bei der Statin-Monotherapie. 0,3 % der Patienten, die die Kombinationsbehandlung erhielten, hatten klinisch relevante Anstiege von Kreatinin auf Werte größer 200 pmol/l.
Die Behandlung sollte abgebrochen werden, wenn der Kreatininwert den oberen Normwert um 50 % übersteigt. Es wird empfohlen, den Kreatininwert während der ersten drei Monate nach Therapiebeginn und danach in periodischen Abständen zu kontrollieren.
Zur Anwendung während Schwangerschaft und Stillzeit siehe Abschnitt 4.6.
4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Orale Antikoagulanzien
Fenofibrat kann die Wirkung von oralen Antikoagulanzien verstärken und folglich ein erhöhtes Blutungsrisiko verursachen. Diese Kombination wird daher nicht empfohlen. Wird eine Behandlung dennoch als notwendig erachtet, sollte zu Beginn der Therapie die Dosis des Antikoagulans um ca. ein Drittel reduziert werden und unter Kontrolle der Gerinnungsparameter (international normalized ratio) - falls erforderlich - angepasst werden.
Ciclosporin
In Einzelfällen wurde über eine erhebliche, wenn auch reversible Einschränkung der Nierenfunktion bei gleichzeitiger Anwendung von fibrathaltigen Arzneimitteln und Ciclosporin berichtet. Daher ist bei diesen Patienten die Nierenfunktion sorgfältig zu überwachen und bei diesbezüglich bedeutsamen Veränderungen der labordiagnostischen Parameter ist Fenofibrat abzusetzen.
HMG-CoA-Reduktase-Hemmer und andere Fibrate
Das Risiko einer ernsthaften Muskelschädigung ist erhöht, wenn ein Fibrat zusammen mit HMG-CoA-Reduktase-Hemmern oder anderen Fibraten kombiniert wird. Eine solche Kombinationstherapie sollte mit Vorsicht eingesetzt und die Patienten sorgfältig auf Anzeichen einer Muskelschädigung hin überwacht werden (siehe Abschnitt 4.4).
Glitazone
Bei gleichzeitiger Einnahme von Fenofibrat und einem Glitazon sind einige Fälle reversibler paradoxer HDL-Cholesterol-Senkung beobachtet worden. Aus diesem Grund sollte das HDL-Cholesterol überwacht werden, wenn einer dieser genannten Wirkstoffe mit dem anderen kombiniert wird. Sollte das HDL-Cholesterol zu niedrig sein, wird empfohlen, eine der beiden Therapien zu beenden.
Cvtochrom-P450-Enzvme
In-vitro-Studien an menschlichen Lebermikrosomen zeigen, dass Fenofibrat und Fenofibrinsäure die Cytochrom-P(CYP-)450-Isoformen CYP3A4, CYP2D6, CYP2E1 oder CYP1A2 nicht inhibieren. Sie sind in therapeutischen Konzentrationen schwache Inhibitoren von CYP2C19 sowie CYP2A6 und mäßige Inhibitoren von CYP2C9.
Patienten, die neben Fenofibrat weitere Arzneimittel mit einer geringen therapeutischen Breite einnehmen, welche über CYP2C19, CYP2A6 und vor allem CYP2C9 metabolisiert werden, sollten sorgfältig überwacht werden. Falls erforderlich, ist die Dosierung dieser Arzneimittel anzupassen.
4.6 Fertilität, Schwangerschaft und Stillzeit
Schwangerschaft
Bei klinischer Anwendung wurden bisher keine Fehlbildungen oder embrvotoxischen Wirkungen beobachtet. Die klinischen Daten reichen jedoch nicht aus, um eine seriöse Abschätzung des Risikos bei Einnahme von Fenofibrat während der Schwangerschaft vornehmen zu können. Tierexperimentelle Studien lassen nicht auf teratogene Wirkungen von Fenofibrat schließen.
Es gibt keine Indikation zum Verordnen von Fibraten während der Schwangerschaft mit Ausnahme von starker Hypertriglyceridämie (> 10 g/l), die durch Diät nur unzureichend behoben werden kann und die Mutter dem Risiko einer akuten Pankreatitis aussetzt.
Stillzeit
Es liegen keine Informationen über die Ausscheidung von Fenofibrat und/oder seinen Metaboliten in die Muttermilch vor. Die Verordnung des Arzneimittels während der Stillzeit wird grundsätzlich nicht empfohlen.
4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
CiL 160 mg hat keinen oder vemachlässigbaren Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
4.8 Nebenwirkungen
Die am häufigsten berichteten unerwünschten Wirkungen unter einer Fenofibrat-Behandlung sind Verdauungsstörungen bzw. gastrointestinale Beschwerden.
Die folgenden Nebenwirkungen wurden im Rahmen placebokontrollierter Studien (n = 2.344) und nach Markteinführung1 mit den angegebenen Häufigkeiten beobachtet:
|
MedDRA- Systemorganklas sen |
Häufig (> 1/100 bis < 1/10) |
Gelegentlich (> 1/1.000 bis < 1/100) |
Selten (> 1/10.000 bis < 1/1.000) |
Nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar) |
|
Erkrankungen des Blutes und des Lymphsystems |
Abnahme von Hämoglobin, Abnahme der Leukozytenzahl | |||
|
Erkrankungen des Immunsystems |
Überempfindlich keit |
Angioödem1 | ||
|
Erkrankungen des Nervensystems |
Kopfschmerzen |
Vertigo | ||
|
Gefäßerkrank ungen |
Thromboembolie (Lungenembolie, tiefe Beinvenenthrom- bose)2 | |||
|
Erkrankungen der Atemwege, des Brustraums und des Mediastinums |
interstitielle Lungen- erkrankungen1 | |||
|
Erkrankungen des Gastrointestinaltrakts |
gastrointestinale Anzeichen und Symptome (Bauchschmerzen, Übelkeit, Erbrechen, Diarrhö, Flatulenz) |
Pankreatitis2 | ||
|
Leber- und Gallenerkrankung en |
Anstieg der Transaminasen (siehe Abschnitt 4.4) |
Cholelithiasis (siehe Abschnitt 4.4) |
Hepatitis (siehe Abschnitt 4.4) |
Ikterus1, Komplikationen einer Cholelithiasis (z. B. Cholezystitis, Cholangitis, Gallenkolik)1 |
|
Erkrankungen der Haut und des Unterhautzellgew |
Überempfindlichkeit der Haut (z. B. |
Alopezie, Photosensibilität |
schwere Hautreaktionen (z. B. Erythema |
|
ebes |
Hautrötungen, Pruritus, Urtikaria) |
multiforme, Steven-JohnsonSyndrom, toxisch epidermale Nekrolyse)1 | ||
|
Skelettmuskulatur -, Bindegewebs-und Knochenerkrank ungen |
Muskelerkrankungen (z. B. Myalgie, Myositis, Muskelkrämpfe und -schwäche) |
Rhabdomyolyse1 | ||
|
Erkrankungen der Geschlechtsorgane und der Brustdrüse |
Potenzstörungen | |||
|
Allgemeine Erkrankungen und Beschwerden am Verarbreichungso rt |
Fatigue | |||
|
Untersuchungen |
Anstieg von Kreatinin im Blut |
Anstieg von Harnstoff im Blut |
2In der FIELD-Studie, einer randomisierten, placebokontrollierten klinischen Untersuchung an 9.795 Patienten mit Typ-2-Diabetes mellitus, wurde bei Patienten, die Fenofibrat erhalten haben, im Vergleich zu Patienten, die Placebo erhalten haben, ein statistisch signifikanter Anstieg an Pankreatitisfällen beobachtet (0,8 % zu 0,5 %; p = 0,031). In derselben Studie wurde von einem statistisch signifikanten Anstieg beim Auftreten von Lungenembolien (0,7 % in der Placebogruppe zu
1,1 % in der Fenofibratgruppe; p = 0,022) und einem statistisch nicht signifikanten Anstieg von Fällen mit tiefer Beinvenenthrombose (Placebo 1 % [48/4.900 Patienten] gegenüber Fenofibrat 1,4 % [67/4.895 Patienten]; p = 0,074) berichtet.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit.
Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem
Bundesinstitut für Arzneimittel und Medizinprodukte Abt. Pharmakovigilanz Kurt-Georg-Kiesinger-Allee 3 D-53175 Bonn Website: www.bfarm.de
anzuzeigen.
4.9 Überdosierung
Nur vereinzelte Fälle von Fenofibrat-Überdosierungen wurden bisher gemeldet. In der Mehrzahl der Fälle wurden keine Überdosierungssymptome berichtet. Ein spezielles Antidot besteht nicht.
Bei Verdacht auf Überdosierung ist symptomatisch zu behandeln und geeignete unterstützende Maßnahmen sind zu ergreifen.
Fenofibrat ist nicht hämodialysierbar.
5. PHARMAKOLOGISCHE EIGENSCHAFTEN
5.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Mittel, die den Lipidstoffwechsel beeinflussen, Fibrate, ATC-Code: C10 AB 05.
Fenofibrat ist ein Derivat der Fibrinsäure, deren lipidregulierende Effekte beim Menschen auf einer Aktivierung der PPARa (Peroxisome Proliferator Activated Rezeptor Type Alpha) beruhen.
Durch die Aktivierung von PPARa wird die Aktivität der Lipoproteinlipase erhöht und die Bildung von Apolipoprotein CIII vermindert. Über diesen Mechanismus steigert Fenofibrat die Lipolyse und Elimination atherogener, triglyceridreicher Partikel aus dem Plasma. Weiterhin wird durch die Aktivierung von PPARa die Synthese der Apolipoproteine AI und AII gesteigert.
Die oben aufgeführten Wirkungen von Fenofibrat führen zu einer Reduktion der Very-low-density-und der Low-density-Lipoproteine (VLDL und LDL), die Apolipoprotein B enthalten, und über eine vermehrte Bildung von Apo AI und Apo AII zu einem Anstieg der High-density-Lipoproteine (HDL).
Patienten mit erhöhtem KHK-Risiko weisen häufig einen atherogenen Lipoprotein-Phänotyp auf, der durch einen erhöhten Anteil an Small-dense-LDL-Partikeln charakterisiert ist. Durch Regulierung der Synthese und des Katabolismus von VLDL senkt Fenofibrat den Small-dense-LDL-Spiegel und erhöht die LDL-Clearance.
In klinischen Studien mit Fenofibrat wurden das Gesamtcholesterin um bis zu 20-25 % und die Triglyceride um 40-55 % gesenkt und HDL-Cholesterin um 10-30 % erhöht.
Bei Patienten mit Hypercholesterinämie, bei denen LDL-Senkungen von 20-35 % beobachtet wurden, führt der Gesamteffekt auf Cholesterin (LDL, HDL) zu einer Senkung des Gesamtcholesterin/HDL-Cholesterin-, des LDL-Cholesterin/HDL-Cholesterin- bzw. des Apo-B/Apo-AI-Quotienten. Die genannten Quotienten gelten als Marker für das atherogene Risiko.
Es liegen Belege dafür vor, dass die Behandlung mit Fibraten die Häufigkeit von Ereignissen bei koronaren Herzerkrankungen reduziert. Es liegen jedoch keine Hinweise für einen positiven Effekt im Hinblick auf die Gesamtmortalität in der primären oder sekundären Vorbeugung kardiovaskulärer Erkrankungen vor.
Bei der ACCORD-(Action to Control Cardiovascular Risk in Diabetes-)Lipid-Studie handelte es sich um eine randomisierte, placebokontrollierte Studie bei 5.518 Patienten mit Typ-2-Diabetes mellitus, die zusätzlich zu Simvastatin mit Fenofibrat behandelt wurden. Bei der Behandlung mit Fenofibrat plus Simvastatin wurden gegenüber der Simvastatin-Monotherapie keine signifikanten Unterschiede hinsichtlich des kombinierten primären Endpunkts, bestehend aus nicht tödlichem Myokardinfarkt, nicht tödlichem Schlaganfall und kardiovaskulär bedingtem Tod, beobachtet (Hazard Ratio [HR] 0,92; 95 %-KI: 0,79-1,08; p = 0,32; absolute Risikoreduktion: 0,74 %). In der vorab festgelegten Untergruppe dyslipidämischer Patienten, definiert als diejenigen Patienten in der untersten Tertile des HDL-C-Werts (< 34 mg/dl bzw. 0,88 mmol/l) und in der obersten Tertile des TG-Werts (> 204 mg/dl bzw. 2,3 mmol/l), wurde bei der Behandlung mit Fenofibrat plus Simvastatin gegenüber der Simvastatin-Monotherapie eine relative Risikoreduktion von 31 % in Bezug auf das kombinierte primäre Zielkriterium beobachtet (Hazard Ratio [HR] 0,69; 95 %-KI: 0,49-0,97; p = 0,03; absolute Risikoreduktion: 4,95 %). Eine weitere vorab festgelegte Untergruppenanalyse ergab eine statistisch signifikante geschlechtsspezifische Interaktion bei der Behandlung (p = 0,01), die auf einen möglichen Behandlungsnutzen der Kombinationstherapie bei Männern hinweist (p = 0,037), während bei Frauen für die Kombinationstherapie im Vergleich zur Simvastatin-Monotherapie ein potenziell höheres Risiko für das Erreichen des primären Endpunkts bestand (p = 0,069). In der bereits genannten Untergruppe dyslipidämischer Patienten wurde eine
7
solche Interaktion nicht beobachtet, es gab jedoch keine klaren Belege für den Nutzen einer Behandlung dyslipidämischer Frauen mit Fenofibrat plus Simvastatin; ferner konnte in dieser Untergruppe eine mögliche nachteilige Wirkung nicht ausgeschlossen werden.
Extravaskuläre Cholesterinablagerungen (Sehnenxanthome und tuberöse Xanthome) können sich während einer Fenofibrat-Therapie teilweise oder vollständig zurückbilden.
Bei Patienten mit erhöhten Lp(a)- bzw. Fibrinogen-Ausgangswerten zeigte sich unter der Behandlung mit Fenofibrat eine signifikante Senkung der Lp(a)- bzw. Fibrinogen-Spiegel. Andere Marker einer Entzündung, wie z. B. C-reaktives Protein, werden unter Fenofibrat ebenfalls reduziert.
Fenofibrat bewirkt eine Reduktion des Harnsäurespiegels um etwa 25 %. Dies ist von zusätzlichem Nutzen für Fettstoffwechselpatienten mit Hyperurikämie.
Fenofibrat führte in tierexperimentellen sowie in einer klinischen Studie zu einer Hemmung der durch ADP, Arachidonsäure und Adrenalin induzierten Thrombozytenaggregation.
5.2 Pharmakokinetische Eigenschaften
CiL 160 mg ist eine Hartkapsel, die 160 mg dispergiertes Fenofibrat enthält, und ist im Vergleich zu bisherigen Darreichungsformen suprabioverfügbar (erhöhte Bioverfügbarkeit).
Resorption
Maximale Plasmaspiegel (Cmax) werden 4-5 Stunden nach oraler Gabe erreicht. Bei wiederholter Applikation bleiben die Plasmakonzentrationen bei allen Patienten konstant.
Die Resorption von Fenofibrat wird durch eine gleichzeitige Nahrungsaufnahme verbessert.
Verteilung
Fenofibrinsäure liegt in hohem Maße (> 99 %) an Albumin gebunden vor.
Biotransformation und Elimination
Nach oraler Gabe wird Fenofibrat schnell durch Esterasen zu dem aktiven Metaboliten Fenofibrinsäure hydrolisiert. Unverändertes Fenofibrat lässt sich im Plasma nicht nachweisen. Fenofibrat ist kein Substrat für CYP3A4. Es ist kein hepatischer mikrosomaler Metabolismus beteiligt.
Der Arzneistoff wird vorwiegend renal und innerhalb von 6 Tagen nahezu vollständig ausgeschieden. Fenofibrat wird hauptsächlich in Form von Fenofibrinsäure und deren Glukuronid eliminiert. Bei älteren Patienten ist die Plasmaausscheidung von Fenofibrinsäure nicht verändert.
Pharmakokinetische Studien mit Einmal- und wiederholter Gabe belegten, dass der Arzneistoff nicht kumuliert. Fenofibrinsäure ist nicht hämodialysierbar.
Die Plasmaeliminationshalbwertszeit von Fenofibrinsäure beträgt etwa 20 Stunden.
Bioverfügbarkeit
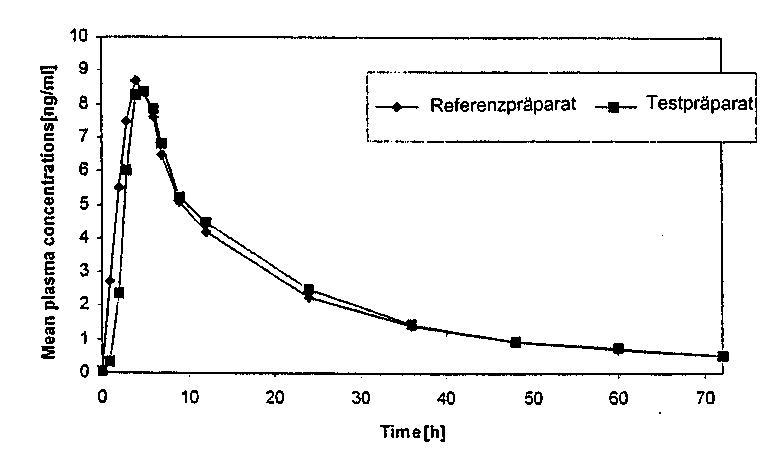
Eine im Jahr 2002 durchgeführte Bioverfügbarkeitsuntersuchung an 18 Probanden ergab im Vergleich zum Referenzpräparat:
Testpräparat
Referenzpräparat
|
Maximale Plasmakonzentration (Cmax) pg/ml |
9,12 ± 2,1 |
9,54 ± 1,97 |
|
Zeitpunkt der maximalen Plasmakonzentration (tmax) h |
4,5 ± 0,98 |
4,17 ± 1,38 |
|
Fläche unter der Kon-zentrations-Zeit-Kurve (AUC) pg x h/ml |
172,43 ± 85,78 |
173,29 ± 90,82 |
Angabe der Werte als Mittelwert und Streubreite.
Mittlere Plasmaspiegelverläufe im Vergleich zu einem Referenzpräparat in einem KonzentrationsZeit-Diagramm:
5.3 Präklinische Daten zur Sicherheit
Untersuchungen zur chronischen Toxizität ergaben keine relevanten Hinweise auf eine spezifische Toxizität von Fenofibrat.
Untersuchungen zur Mutagenität von Fenofibrat verliefen negativ.
Bei Ratten und Mäusen wurden in hohen Dosierungen Lebertumore gefunden, die auf Peroxisomenproliferation zurückzuführen sind. Diese Veränderungen sind spezifisch für kleine Nager und wurden bei anderen Tierarten nicht beobachtet. Eine Relevanz für die therapeutische Anwendung beim Menschen ergibt sich daraus nicht.
Untersuchungen an Maus, Ratte und Kaninchen ergaben keine Hinweise auf eine teratogene Wirkung. Embryotoxische Effekte wurden bei Dosierungen, die im maternaltoxischen Bereich lagen, beobachtet. In hohen Dosen traten Tragzeitverlängerungen und eine Beeinträchtigung des Geburtsvorganges auf.
Hinweise auf eine Beeinflussung der Fertilität ergaben sich nicht.
6. PHARMAZEUTISCHE ANGABEN
6.1 Liste der sonstigen Bestandteile
Macrogolglycerollaurate (Ph. Eur.), Macrogol 20.000, Hyprolose, Carboxymethylstärke-Natrium (Typ A) (Ph. Eur.).
Kapselhülle: Gelatine, Eisen(n,III)-oxid, Eisen(III)-hydroxid-oxid, Titandioxid, Indigocarmin.
6.2 Inkompatibilitäten Nicht zutreffend.
6.3 Dauer der Haltbarkeit 3 Jahre.
Dieses Arzneimittel soll nach Ablauf des Verfalldatums nicht mehr angewendet werden.
6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Für dieses Arzneimittel sind keine besonderen Lagerungsbedingungen erforderlich.
6.5 Art und Inhalt des Behältnisses
Aluminium/PVC-Blisterpackungen in einer Faltschachtel.
Packungen mit 30, 50 und 100 Hartkapseln.
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung
Keine besonderen Anforderungen.
7. INHABER DER ZULASSUNG
Winthrop Arzneimittel GmbH 65927 Frankfurt am Main
Mitvertrieb
Zentiva Pharma GmbH
65927 Frankfurt am Main Telefon: (01 80) 2 02 00 101
Telefax: (01 80) 2 02 00 111
8. ZULASSUNGSNUMMER
54880.00.00
9. DATUM DER ERTEILUNG DER ZULASSUNG/VERLÄNGERUNG DER ZULASSUNG
Datum der Erteilung der Zulassung: 6. April 2004
Datum der letzten Verlängerung der Zulassung: 30. April 2009
10. STAND DER INFORMATION
September 2016
11. VERKAUFSABGRENZUNG
Verschreibungspflichtig. 1
11
Mat.-Nr.: 325973
0,06 €/Anruf (dt. Festnetz); max. 0,42 €/min (Mobilfunk).