Cluvot 1250 I.e.
CSL Behring
Cluvot®
GEBRAUCHSINFORMATION UND FACHINFORMATION
Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden. Hinweise zur Meldung von Nebenwirkungen, siehe Abschnitt 4.8.
1. BEZEICHNUNG DES ARZNEIMITTELS
Cluvot 250 I.E., Pulver und Lösungsmittel zur Herstellung einer Injektions-/Infusionslösung Cluvot 1250 I.E., Pulver und Lösungsmittel zur Herstellung einer Injektions-/Infusionslösung
2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
Wirkstoff: Cluvot ist ein gereinigtes Konzentrat des aus menschlichen Plasma gewonnenen Gerinnungsfaktors XIII (FXIII). Es liegt als weißes Pulver vor.
Jede Durchstechflasche enthält nominal 250 oder 1250 I.E. humanen Blutgerinnungsfaktor XIII. Cluvot enthält ungefähr 62,5 I.E./ml (250 I.E./4 ml und 1250 I.E./20 ml) humanen Blutgerinnungsfaktor XIII, wenn man es mit 4 bzw. 20 ml Wasser für Injektionszwecke rekonstituiert.
Die spezifische Aktivität von Cluvot beträgt ungefähr 6 - 10 I.E./mg Protein.
Vollständige Auflistung der sonstigen Bestandteile siehe Abschnitt 6.1.
3. DARREICHUNGSFORM
Pulver und Lösungsmittel zur Herstellung einer Injektions-/Infusionslösung.
Weißes Pulver und klare, farblose Lösung.
4. KLINISCHE ANGABEN
4.1 Anwendungsgebiete
Cluvot ist indiziert für Erwachsene und Kinder
• für die Prophylaxe bei kongenitalem Mangel an FXIII und
• für die perioperative Behandlung von chirurgischen Blutungen bei kongenitalem FXIII Mangel.
4.2 Dosierung und Art der Anwendung
Dosierung
1 ml entspricht ungefähr 62,5 I.E. bzw. 100 I.E. entsprechen 1,6 ml.
Wichtig:
Dosierung und Dauer der Therapie sollten sich stets an der klinischen Wirksamkeit im
Einzelfall orientieren.
Verabreichung
Die Dosierung sollte individuell auf das Körpergewicht, die Laborwerte und das
Krankheitsbild des Patienten ausgerichtet werden.
Dosierung zur Routineprophylaxe
Initiale Dosis
• 40 internationale Einheiten (I.E.) pro kg Körpergewicht
• Die Injektionsrate sollte 4 ml pro Minute nicht überschreiten.
Anschließende Dosierung
• Die Dosierung sollte sich an dem letzten FXIII-Aktivität-Talspiegel orientieren. Das Intervall beträgt 28 Tage (4 Wochen), um einen FXIII-Aktivität-Talspiegel von ungefähr 5 bis 20 % aufrecht zu erhalten
• Empfohlene Dosisanpassungen von + 5 I. E. pro kg sollten die in Tabelle 1 gezeigten FXIII-Aktivität-Talspiegel und das Krankheitsbild des Patienten berücksichtigen.
• Dosisanpassungen sollten auf der Grundlage einer speziellen sensitiven Untersuchung gemacht werden, die benutzt wird, um den FXIII-Spiegel zu bestimmen. Ein Beispiel der Dosisanpassung mit Hilfe der Standard Berichrom® Aktivitäts-Messung wird unten in Tabelle 1 gezeigt.
Tabelle 1: Dosisanpassung mit Hilfe der Berichrom® Aktivitäts-Messung
|
FXIII-Aktivität-Talspiegel (%) |
Dosisänderung |
|
Ein Talspiegel von < 5% |
Anstieg um 5 Einheiten pro kg |
|
Talspiegel von 5 % bis 2 0% |
Keine Änderung |
|
Zwei Talspiegel von > 20 % |
Abnahme um 5 Einheiten pro kg |
|
Ein Talspiegel von >25 % |
Abnahme um 5 Einheiten pro kg |
Die in Einheiten gemessene Stärke wird mit Hilfe der Berichrom® Aktivitäts-Messung bestimmt und bezieht sich auf den aktuellen internationalen Standard für Blutgerinnungsfaktor XIII, Plasma. Daher ist eine Einheit gleichzusetzen mit einer internationalen Einheit.
Präoperative Prophylaxe
Nach der letzten Routineprophylaxe des Patienten, wenn eine Operation geplant ist:
• Zwischen 21 und 28 Tage danach - es wird eine volle Prophylaxe-Dosis direkt vor der Operation verabreicht. Die nächste Prophylaxe-Dosis sollte nach 28 Tagen gegeben werden.
• Zwischen 8 und 21 Tage danach - es kann eine zusätzliche Dosis (voll oder teilweise) vor der Operation verabreicht werden. Die Dosis sollte sich an dem FXIII-Aktivitäts-Spiegel und dem Krankheitsbild des Patienten orientieren und sollte an die Halbwertszeit von Cluvot angepasst werden.
• Innerhalb von 7 Tagen nach der letzten Dosis - zusätzliche Gabe wird möglicherweise nicht nötig sein.
Dosisanpassungen können von diesen Empfehlungen abweichen und sollten sich stets am FXIII-Spiegel und dem Krankheitsbild des Patienten orientieren. Alle Patienten sollten während und nach der Operation engmaschig überwacht werden.
Daher empfiehlt es sich, den Anstieg der FXIII-Aktivität mit einer FXIII-Bestimmung zu überwachen. Bei größeren Eingriffen und schweren Blutungen sind annähernde Normalwerte anzustreben (gesunde Personen: 70 % - 140 %).
Kinder und Jugendliche
Die Dosierung und Art der Anwendung bei Kindern und Jugendlichen basiert auf dem Körpergewicht und entspricht damit im Allgemeinen denselben Richtlinien wie für Erwachsene. Als Grundlage für die Dosierung und/oder Häufigkeit der Anwendung sollte immer die klinische Effektivität und der FXIII-Aktivitätsspiegel dienen.
Ältere Patienten
Die Dosierung und Art der Anwendung bei älteren Personen (> 65 Jahren) wurde nicht in klinischen Studien untersucht.
Art der Anwendung
Nach der Rekonstitution sollte die Lösung klar oder leicht opaleszent sein. Die gebrauchsfertige Lösung soll vor der Anwendung auf Raum- oder Körpertemperatur angewärmt werden und langsam intravenös mit einer für den Patienten angenehmen Geschwindigkeit injiziert oder infundiert werden. Die Injektions- oder Infusionsgeschwindigkeit soll ca. 4 ml pro Minute nicht überschreiten.
Der Patient soll auf sofortige Reaktionen beobachtet werden. Wenn eine Reaktion erfolgt, die mit der Verabreichung von Cluvot in Zusammenhang gebracht werden könnte, soll - in Abhängigkeit vom klinischen Zustand des Patienten - die Infusionsgeschwindigkeit gesenkt bzw. die Infusion abgebrochen werden.
Hinweise zur Rekonstitution des Arzneimittels vor der Anwendung, siehe Abschnitt 6.6.
4.3 Gegenanzeigen
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Bei Patienten mit bekannten allergischen Reaktionen auf das Präparat (mit Symptomen wie generalisierter Nesselsucht, Hautrötung, Blutdruckabfall, Atembeschwerden) können Antihistaminika und Kortikosteroide vorbeugend verabreicht werden.
Allergische Überempfindlichkeitsreaktionen bei der Anwendung von Cluvot sind möglich. Falls Symptome einer Überempfindlichkeit (wie quaddelartiger Hautausschlag, generalisierte Nesselsucht, Engegefühl in der Brust, Stridor, Blutdruckabfall und Anaphylaxie) auftreten, sollte die Cluvot-Infusion sofort gestoppt werden. Bei einem Schock sollen die aktuellen medizinischen Richtlinien zur Schockbehandlung beachtet werden.
Bei frischen Thrombosen ist wegen der fibrinstabilisierenden Wirkung Vorsicht geboten. Immunogenität
Die Bildung neutralisierender Antikörper (Inhibitoren) gegen FXIII ist eine bekannte Komplikation bei der Behandlung mit Cluvot. Daher sollten Patienten auf mögliche Entwicklung von Inhibitoren überwacht werden. Das Vorhandensein von Inhibitoren kann sich als nicht ausreichendes Ansprechen auf die Therapie darstellen. Bei Verdacht auf Inhibitoren, bei nicht erreichten Plasma FXIII-Spiegeln, oder bei Blutungen während der Prophylaxebehandlung sollte die FXIII-Inhibitor Konzentration gemessen werden.
Hinweis für Patienten mit salzarmer Diät
Cluvot enthält 124,4 - 195,4 mg (5,41 - 8,50 mmol) Natrium pro Dosis (40 I.E./Körpergewicht bei durchschnittlich 70 kg), wenn die empfohlene Dosis (2800 I.E. = 44,8 ml) verabreicht wird. Dies sollte bei Patienten berücksichtigt werden, die eine salzarme Diät einhalten müssen.
Virussicherheit
Standardmethoden zur Vermeidung von Infektionskrankheiten, die im Rahmen der Anwendung von aus menschlichem Blut oder Plasma hergestellten Arzneimitteln auftreten können, umfassen die Auswahl der Spender, die Prüfung jeder einzelnen Spende und jedes Plasmapools auf spezifische Marker für Infektionen sowie die Einbeziehung effektiver Herstellungsschritte zur Inaktivierung/Eliminierung von Viren.
Trotz dieser Maßnahmen kann die Möglichkeit der Übertragung von Erregern bei der Anwendung von aus menschlichem Blut oder Plasma hergestellten Arzneimitteln nicht vollständig ausgeschlossen werden. Dies gilt auch für bisher unbekannte Viren und andere Pathogene.
Die getroffenen Maßnahmen werden als wirksam angesehen für umhüllte Viren, wie z. B. das humane Immundefizienzvirus (HIV), das Hepatitits B-Virus (HBV) und das Hepatitis C-Virus (HCV) sowie für die nicht-umhüllten Viren Hepatitis A-Virus (HAV) und Parvovirus B19.
Es wird auf die Dokumentationspflicht gemäß Transfusionsgesetz hingewiesen.
Für Patienten, die regelmäßig Präparate aus menschlichem Blut oder Plasma erhalten, wird eine Impfung gegen Hepatitis A und Hepatitis B empfohlen.
4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Es wurden keine Studien zur Erfassung von Wechselwirkungen durchgeführt.
4.6 Fertilität, Schwangerschaft und Stillzeit Schwangerschaft
Eingeschränkte Daten zur klinischen Anwendung von Cluvot in der Schwangerschaft zeigten keine negativen Auswirkungen auf den Schwangerschaftsverlauf oder die Entwicklungsphase während oder nach der Geburt.
Die Anwendung von Cluvot in der Schwangerschaft kann, wenn nötig, in Betracht gezogen werden.
Stillzeit
Es gibt keine Daten über die Ausscheidung von Cluvot in die Muttermilch. Aufgrund seiner Molekülgröße ist die Ausscheidung in die Milch jedoch unwahrscheinlich und - aufgrund seiner proteinartigen Struktur - die Absorption von ganzen Molekülen in den kindlichen Körper auch unwahrscheinlich. Daher kann Cluvot während der Stillzeit verabreicht werden.
Fertilität
Es gibt keine Daten über die Auswirkungen von Cluvot auf die Fortpflanzungsfähigkeit.
4.7 Auswirkung auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Es wurden keine Studien zu den Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen durchgeführt.
4.8 Nebenwirkungen
Die im folgenden genannten Nebenwirkungen beruhen auf Analysen von Post-MarketingDaten.
Tabellarische Auflistung der Nebenwirkungen
Die unten stehende Tabelle ist eingeteilt nach der MedDRA-Systemorganklassifikation.
Die folgenden Standard-Kategorien von Häufigkeiten werden verwendet:
Sehr häufig (> 1/10); häufig (> 1/100 bis < 1/10); gelegentlich (> 1/1.000 bis < 1/100); selten (> 1/10.000 bis < 1/1.000); sehr selten (< 1/10.000).
|
MedDRA Systemorganklasse |
Unerwünschte Wirkung |
Häufigkeit |
|
Erkrankungen des Immunsystems |
Allergoi d-anaphylaktoide Reaktionen (wie generalisierte Nesselsucht, Hautrötung, Blutdruckabfall, Atembeschwerden) |
Selten |
|
Entstehung von Inhibitoren gegen FXIII |
Sehr selten | |
|
Allgmeine Erkrankungen und Beschwerden am Verabreichungsort |
Temperaturanstieg |
Selten |
Bei Auftreten von allergoid-anaphylaktoiden Reaktionen ist Cluvot sofort abzusetzen und eine situationsgerechte Behandlung einzuleiten. Die aktuellen medizinischen Richtlinien zur Schockbehandlung sind zu beachten.
Kinder und Jugendliche:
Das Sicherheitsprofil für Kinder und Jugendliche in klinischen Studien entspricht dem von Erwachsenen.
Informationen zur Virussicherheit siehe Abschnitt 4.4.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-RisikoVerhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel, Paul-Ehrlich-Institut, Paul-Ehrlich-Str. 51-59, 63225 Langen, Telefon: +49 6103 770, Telefax: +49 6103 77 1234, Webseite: www.pei.de anzuzeigen.
4.9 Überdosierung
Es wurden keine Fälle von Überdosierung berichtet.
5. PHARMAKOLOGISCHE EIGENSCHAFTEN
5.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Antihämorrhagika ATC-Code: B02B D07
Da Faktor XIII enzymatisch Aminogruppen des Lysin mit Glutamin verbindet, ist er in der Lage, Fibrinmoleküle miteinander zu vernetzen (Transamidase-Wirkung). Dies hat zur
Folge, dass Gerinnsel stabilisiert werden und Fibroblasten beschleunigt in die Gerinnsel einsprossen, so dass die Wundheilung gefördert wird.
Kinder und Jugendliche
In klinischen Studien, an denen auch Kinder und Jugendliche < 18 Jahre mit kongenitalem FXIII-Mangel teilnahmen, wurde gezeigt, dass bei prophylaktischer Anwendung von Cluvot alle 28 Tage ein FXIII-Aktivität-Talspiegel von ca. 5 % bis 20 % erreicht werden kann.
5.2 Pharmakokinetische Eigenschaften
Verteilung
Das Präparat wird intravenös appliziert und ist sofort in einer der Dosierung entsprechenden Plasmakonzentration verfügbar.
Elimination
Die biologische Halbwertszeit wurde bei Patienten mit kongenitalem FXIII-Mangel mit 6,6 ± 2,29 Tagen (Median ± SD) bestimmt. Cluvot wird wie der körpereigene Gerinnungsfaktor XIII abgebaut.
Die folgende Tabelle gibt einen Überblick über die pharmakokinetischen Parameter (Erwachsene/18 Jahre und älter):
|
Parameter |
Median (min -max) |
|
AUC ss, 0-inf (Einheiten»h/ml) |
182,9 (133,5 - 300,2) |
|
Css, max (Einheiten/ml)* |
0,9 (0,6-1,2) |
|
Css, min (Einheiten/ml)* |
0,07 (0,0 - 0,16) |
|
Tmax (h) |
1,2 (0,7 - 4,2) |
|
Halbwertszeit [Tage] |
7,8 (3,1 - 11,02) |
|
CL [ml/h/kg] |
0,22 (0,13 - 0,30) |
|
Vss [ml/kg] |
49,4 (31,65 - 62,91) |
|
MRT [Tage] |
11,7 (5,7 - 17,02) |
AUC ss, (o-irf) = Fläche unter der Plasmakonzentrationskurve
von Zeit 0 bis Unendlichkeit bei steady state
* 100 % Aktivität entspricht 1 Einheit/ml
Css, max: Höchste Konzentration bei steady state
Css, min: Niedrigste Konzentration bei steady state
Tmax: Zeit bis zur höchsten Konzentration
CL: Clearance
Vss: Verteilungsvolumen bei steady state MRT = Mittlere Verweildauer
Kinder und Jugendliche
Von den 188 Studienteilnehmern der FXIII Konzentrations-Studien (am Menschen) waren bei Aufnahme 117 Teilnehmer < 18 Jahre (1 Monat bis < 2 Jahre, n = 17; 2 Jahre bis < 12 Jahre, n = 62; 12 Jahre bis < 16 Jahre, n = 30; 17 Jahre bis 18 Jahre, n = 8). In der Pharmakokinetikstudie PK 2002 waren 5 von den 14 Studienteilnehmern zwischen 2 und < 18 Jahre alt (2 bis 11 Jahre, n = 3; 12 bis 16 Jahre, n = 2, 17 bis 18 Jahre, n = 0). Teilnehmer die jünger als 16 Jahre waren, hatten eine kürzere Halbwertszeit und schnellere Clearance (Halbwertszeit: 5,7 ± 1,00 Tage; Clearance: 0,291±0,12 ml/h/kg) im Vergleich zu Erwachsenen (Halbwertszeit: 7,1 ± 2,74 Tage, Clearance: 0,22 ± 0,07 ml/h/kg).
Das Produkt hat bei Kindern eine kürzere Halbwertszeit und schnellere Clearance als bei Erwachsenen. Da jedoch die Dosierung unabhängig von der Altersgruppe individuell nach dem Körpergewicht berechnet und dem FXIII-Aktivität-Talspiegel angepasst wird, ist keine altersgemäße Dosierung nötig.
5.3 Präklinische Daten zur Sicherheit
Die in Cluvot enthaltenen humanen Plasmaproteine verhalten sich wie körpereigene Bestandteile.
Die Untersuchungen an Labortieren mit einmaliger und wiederholter Dosierung ergaben keinen Hinweis auf ein toxikologisches Potential von Cluvot.
Reproduktionsstudien und Studien zur embryofötalen Entwicklung wurden nicht durchgeführt.
6. PHARMAZEUTISCHE ANGABEN
6.1 Liste der sonstigen Bestandteile
Pulver:
Human Albumin
Glucosemonohydrat
Natriumchlorid
NaOH (zur Einstellung des pH-Wertes)
Lösungsmittel:
Wasser für Injektionszwecke
6.2 Inkompatibilitäten
Cluvot darf, außer mit den unter Abschnitt 6.6 aufgeführten, nicht mit anderen Arzneimitteln gemischt werden und soll über einen separaten Zugang verabreicht werden.
6.3 Dauer der Haltbarkeit
3 Jahre.
Cluvot darf nach Ablauf des auf der Packung und dem Behältnisses angegebenen Verfallsdatums nicht mehr angewendet werden.
Die physiko-chemische Stabilität ist für 24 Stunden bei < 25 °C belegt. Allerdings sollte das Präparat aus mikrobiologischer Sicht sofort verwendet werden. Wenn das Produkt nicht sofort verbraucht wird, darf es nicht länger als 4 Stunden bei Raumtemperatur gelagert werden. Das gelöste Produkt darf nicht im Kühlschrank aufbewahrt oder eingefroren werden.
6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Im Kühlschrank lagern (+ 2 °C bis + 8 °C).
Nicht einfrieren.
Die Durchstechflaschen in der geschlossenen Faltschachtel aufbewahren, um den Inhalt vor Licht zu schützen.
Für Hinweise zur Aufbewahrung nach Rekonstitution des Produkts siehe Abschnitt 6.3.
6.5 Art und Inhalt der Behältnisse
Durchstechflaschen:
250 I.E.
Pulver: Durchstechflasche aus farblosem Glas, verschlossen mit einem Gummistopfen (Bromobutyl) und versiegelt mit einer Aluminium/Kunststoff-Bördelkappe.
Lösungsmittel (Wasser für Injektionszwecke): Durchstechflasche aus farblosem Glas 1250 I.E.
Pulver: Durchstechflasche aus farblosem Glas, verschlossen mit einem Gummistopfen (Bromobutyl) und versiegelt mit einer Aluminium/Kunststoff-Bördelkappe.
Lösungsmittel (Wasser für Injektionszwecke): Durchstechflasche aus farblosem Glas.
Packungsgrößen Packung mit 250 I.E.
1 Durchstechflasche mit Pulver
1 Durchstechflasche mit 4 ml Wasser für Injektionszwecke 1 Filter Transfer Set 20/20 (Mix2Vial)
Packung mit 1250 I.E.
1 Durchstechflasche mit Pulver
1 Durchstechflasche mit 20 ml Wasser für Injektionszwecke 1 Filter Transfer Set 20/20 (Mix2Vial)
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung
Allgemeine Hinweise
Die Lösung sollte klar oder leicht opaleszent sein. Das rekonstituierte Produkt sollte nach der Filtration/Aufziehen der Lösung in die Spritze (siehe unten) und vor der Anwendung auf Partikel und Verfärbungen visuell überprüft werden. Deutlich trübe Lösungen und Lösungen mit Ausflockungen oder Niederschlag dürfen nicht verwendet werden. Zubereitung und Entnahme müssen unter aseptischen Bedingungen erfolgen.
Zubereitung
Erwärmen Sie das Lösungsmittel auf Raumtemperatur. Vor dem Öffnen der Mix2Vial Packung die Flip-Off-Kappen der Lösungsmittel- und Produktflaschen entfernen und die Stopfen mit einer antiseptischen Lösung behandeln und anschließend trocknen lassen.
1
2
3
4
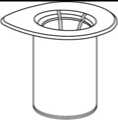
1. Entfernen Sie das Deckpapier von der Mix2Vial Packung. Das Mix2Vial nicht aus dem Blister entnehmen!
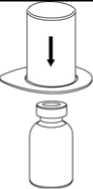
2. Die Lösungsmittelflasche auf eine ebene, saubere Fläche stellen und festhalten. Das Mix2Vial Set mit dem Blister greifen und den Dorn des blauen Adapters senkrecht in den Stopfen der Lösungsmittelflasche einstechen.
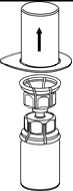
3. Vorsichtig die Verpackung vom Mix2Vial Set entfernen, indem man den Blister am Siegelrand fasst und ihn senkrecht nach oben abzieht. Dabei ist darauf zu achten, dass nur der Blister und nicht das Mix2Vial entfernt wird.
4. Die Produktflasche auf eine feste Unterlage stellen. Die Lösungsmittelflasche mit dem aufgesetzten Mix2Vial Set herumdrehen und den Dorn des transparenten Adapters senkrecht in den Stopfen der Produktflasche einstechen. Das Lösungsmittel läuft automatisch in die Produktflasche über.
5
6
7
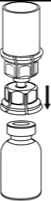
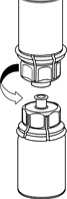
5. Mit der einen Hand die Produktseite und mit der anderen Hand die Lösungsmittelseite des Mix2Vial greifen und das Set vorsichtig auseinander schrauben. Entsorgen Sie die Lösungsmittelflasche mit dem blauen Mix2Vial Adapter.
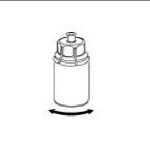
6. Die Produktflasche mit dem transparenten Adapter vorsichtig schwenken, bis das Produkt vollständig gelöst ist. Nicht schütteln.
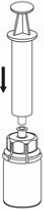
7. Luft in eine leere, sterile Spritze aufziehen. Die Produktflasche aufrecht halten, die Spritze mit dem Luer Lock Anschluss des Mix2Vial Set verbinden und die Luft in die Produktflasche injizieren.
Aufziehen der Lösung in die Spritze und Anwendung
|
l |
“1 P 8 |
8. Den Stempel der Spritze gedrückt halten, das gesamte System herumdrehen und die Lösung durch langsames Zurückziehen der Kolbenstange in die Spritze aufziehen. |
|
1 |
9. Nachdem die Lösung vollständig in die Spritze überführt ist, den Spritzenzylinder fassen (dabei die Kolbenstange in ihrer Position halten) und die Spritze vom transparenten Mix2Vial Adapter abdrehen. |
Es ist darauf zu achten, dass kein Blut in die gefüllte Spritze gelangt, da die Gefahr besteht, dass es dort gerinnt und dadurch dem Patienten Fibringerinnsel verabreicht werden.
Die Lösung soll langsam intravenös (nicht schneller als 4 ml pro Minute) über einen separaten Injektions-/Infusions-Zugang verabreicht werden.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
7. INHABER DER ZULASSUNG
CSL Behring GmbH
- Emil-von-Behring-Str. 76 35041 Marburg
- V erkauf Deutschland Philipp-Reis-Str. 2 65795 Hattersheim Tel.: (069) 305 - 8 44 37 Fax: (069) 305 - 1 71 29
8. ZULASSUNGSNUMMERN
Cluvot® 250 Zul.- Nr.: Cluvot® 1250 Zul.- Nr.:
9. DATUM DER ERTEILUNG DER ZULASSUNG/VERLÄNGERUNG DER ZULASSUNG
Zulassung:
10. STAND DER INFORMATION
Februar 2014
11. HERKUNFTSLÄNDER DES BLUTPLASMAS
Deutschland, Österreich, Polen, USA
12. VERSCHREIBUNGSSTATUS
Verschreibungspflichtig