Convulex 500
FACHINFORMATION
1. BEZEICHNUNG DER ARZNEIMITTEL
Convulex® 150
150 mg/magensaftresistente Kapsel Convulex® 300
300 mg/magensaftresistente Kapsel Convulex® 500
500 mg/magensaftresistente Kapsel Wirkstoff: Valproinsäure
Convulex® Tropflösung
300 mg/1 ml Lösung zum Einnehmen
Wirkstoff: Natriumvalproat
2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
Convulex® 150:
1 magensaftresistente Kapsel enthält 150 mg Valproinsäure Sonstiger Bestandteil: Sorbitol
Convulex® 300:
1 magensaftresistente Kapsel enthält 300 mg Valproinsäure Sonstiger Bestandteil: Sorbitol
Convulex® 500:
1 magensaftresistente Kapsel enthält 500 mg Valproinsäure Sonstiger Bestandteil: Sorbitol
Convulex® Tropflösung:
1 ml Lösung enthält
300 mg Natriumvalproat (entsprechend 260,1 mg Valproinsäure)
Die vollständige Auflistung der sonstigen Bestandteile siehe Abschnitt 6.1.
3. DARREICHUNGSFORM
Convulex® 150 / -300: magensaftresistente Kapsel
Ovale, altrosafarbene magensaftresistente Kapseln mit der Aufschrift 150 bzw. 300 auf einer Seite Convulex® 500: magensaftresistente Kapsel
Oblonge, altrosafarbene magensaftresistente Kapseln mit der Aufschrift 500 auf einer Seite
Convulex® Tropflösung: Lösung zum Einnehmen
Klare, farblose bis leicht gelbstichige Lösung zum Einnehmen
KLINISCHE ANGABEN
4.
4.1 Anwendungsgebiete
Zur Behandlung von:
- Generalisierten Anfällen in Form von Absencen, myoklonischen Anfällen und tonisch-klonischen Anfällen,
- fokalen und sekundär-generalisierten Anfällen
- und zur Kombinationsbehandlung bei anderen Anfallsformen, z. B. fokalen Anfällen mit einfacher und komplexer Symptomatologie sowie fokalen Anfällen mit sekundärer Generalisation, wenn diese Anfallsformen auf die übliche antiepileptische Behandlung nicht ansprechen.
Hinweis:
Bei Kleinkindern sind valproinsäurehaltige Arzneimittel nur in Ausnahmefällen Mittel erster Wahl; Convulex® sollte nur unter besonderer Vorsicht nach strenger Nutzen-Risiko-Abwägung und möglichst als Monotherapie angewendet werden.
4.2 Dosierung, Art und Dauer der Anwendung
Die Dosierung ist individuell vom (Fach)arzt zu bestimmen und zu kontrollieren, wobei Anfallsfreiheit bei minimaler Dosierung, besonders auch in der Schwangerschaft, angestrebt werden sollte.
Es empfiehlt sich ein stufenweiser (einschleichender) Aufbau der Dosierung bis zur optimal wirksamen Dosis.
In der Monotherapie beträgt die Initialdosis in der Regel 5 - 10 mg Valproinsäure (entsprechend 5,8 -11,5 mg Natriumvalproat)/kg Körpergewicht, die alle 4 - 7 Tage um etwa 5 mg Valproinsäure (entsprechend 5,8 mg Natriumvalproat)/kg Körpergewicht erhöht werden sollte.
Die volle Wirkung ist in einigen Fällen erst nach 4 - 6 Wochen zu beobachten. Die Tagesdosen sollen deshalb nicht zu früh über mittlere Werte hinaus gesteigert werden.
Die mittlere Tagesdosis beträgt während der Langzeitbehandlung im Allgemeinen für:
- Erwachsene 20 mg Valproinsäure (entsprechend 23 mg Natriumvalproat)/kg Körpergewicht,
- Jugendliche 25 mg Valproinsäure (entsprechend 28,8 mg Natriumvalproat)/kg Körpergewicht,
- Kinder 30 mg Valproinsäure (entsprechend 34,6 mg Natriumvalproat)/kg Körpergewicht.
Entsprechend werden folgende orientierende Tagesdosen empfohlen:
Dosierungstabellen:
Convulex® 150:
|
Lebensalter |
Körpergewicht (in kg) |
Durchschnittliche ValproinsäureDosis in mg/Tag |
Anzahl von Kapseln |
|
3 - 6 Jahre |
ca. 15 - 25 |
300 - 600 |
2 - 4 |
|
7 - 14 Jahre |
ca. 25 - 40 |
450 - 1500 |
3 - 10 |
Für die Anfangsbehandlung von Säuglingen und Kleinkindern steht außerdem Convulex® Tropflösung zur Verfügung (Dosierungstabelle siehe unten), die auch zur exakten Einstellung mit Convulex® 150 kombiniert werden kann.
Jugendliche und Erwachsene werden im Allgemeinen mit Convulex® 300 und Convulex® 500 Kapseln behandelt.
Convulex® 300:
|
Lebensalter |
Körpergewicht (in kg) |
Durchschnittliche ValproinsäureDosis in mg/Tag |
Anzahl von Kapseln |
|
Erwachsene |
ab ca. 60 |
1200 - 2100 |
4 - 7 |
|
Jugendliche |
ca. 40 - 60 |
600 - 1500 |
2 - 5 |
|
ab 14 Jahre | |||
|
Kinder |
ca. 25 - 40 |
600 - 1200 |
2 - 4 |
|
7 - 14 Jahre |
Für die exakte Einstellung bzw. für die Behandlung von Säuglingen und Kleinkindern stehen außerdem die Darreichungsformen Convulex® 150 und Convulex® Tropflösung zur Verfügung. Zur Behandlung mit höheren Dosen liegt die Darreichungsform Convulex® 500 vor.
Convulex® 500:
|
Lebensalter |
Körpergewicht (in kg) |
Durchschnittliche ValproinsäureDosis in mg/Tag |
Anzahl von Kapseln |
|
Erwachsene |
ab ca. 60 |
1000 - 2000 (- 2500) |
2 - 4 (- 5) |
|
Jugendliche |
ca. 40 - 60 |
1000 - 1500 |
2 - 3 |
|
ab 14 Jahre | |||
|
Kinder |
ca. 25 - 40 |
500 - 1500 |
1 - 3 |
|
7 - 14 Jahre |
Für die exakte Einstellung bzw. für die Behandlung von Säuglingen und Kleinkindern stehen auch die Darreichungsformen Convulex® 150 und Convulex® 300 Kapseln sowie Convulex® Tropflösung zur Verfügung.
Convulex® Tropflösung:
|
Lebensalter |
Körpergewicht (in kg) |
Durchschnittliche Dosis in mg/Tag |
|
3 - 6 Monate |
ca. 5,5 - 7,5 kg |
173 mg Natriumvalproat (entsprechend 150 mg Valproinsäure) |
|
6 - 12 Monate |
ca. 7,5 - 10 kg |
173 - 346 mg Natriumvalproat (entsprechend 150 - 300 mg Valproinsäure) |
|
1 - 3 Jahre |
ca. 10 - 15 kg |
346 - 518 mg Natriumvalproat (entsprechend 300 - 450 mg Valproinsäure) |
|
3 - 6 Jahre |
ca. 15 - 25 kg |
346 - 692 mg Natriumvalproat (entsprechend 300 - 600 mg Valproinsäure) |
Ältere Kinder, Jugendliche und Erwachsene werden im Allgemeinen mit Convulex® 150, Convulex® 300 und Convulex® 500 Kapseln behandelt.
Wird Convulex® in Kombination oder als Substitutionstherapie zu einer früheren Medikation gegeben, muss die Dosis der bis dahin eingenommenen Antiepileptika, besonders des Phenobarbitals, unverzüglich vermindert werden. Falls die vorausgegangene Medikation abgesetzt wird, hat dies ausschleichend zu erfolgen.
Da die enzyminduzierende Wirkung anderer Antiepileptika reversibel ist, ist etwa 4 - 6 Wochen nach der letzten Einnahme eines solchen Antiepileptikums der Serumspiegel der Valproinsäure zu kontrollieren und die Tagesdosis gegebenenfalls zu reduzieren.
Die Serumkonzentration (bestimmt vor der ersten Tagesdosis) sollte 100 pg Valproinsäure/ml nicht überschreiten.
Bei Patienten mit Niereninsuffizienz oder Hypoproteinämie muss der Anstieg an freier Valproinsäure im Serum in Betracht gezogen und die Dosis gegebenenfalls reduziert werden. Entscheidend für eine Dosisanpassung sollte jedoch das klinische Bild sein, da eine Bestimmung der Valproinsäuregesamtkonzentration im Serum zu falschen Schlussfolgerungen führen kann (siehe Abschnitt 5.2).
Die Pharmakokinetik von Valproinsäure kann bei älteren Patienten verändert sein. Die Dosierung sollte anhand der Anfallskontrolle bestimmt werden (siehe Abschnitt 5.2).
Die Tagesdosis kann auf 2 - 4 Einzelgaben verteilt werden.
Folgende Tagesdosen werden empfohlen: siehe Dosierungstabellen.
Convulex® Tropflösung:
Zur genauen Dosierung liegt der Packung eine graduierte Dosierspritze bei.
Die Dosierspritze ist mit einer Dosierungsskala versehen: Die Ziffern auf der Dosierspritze geben die mg-und die ml-Dosierung an. Die mg-Angaben beziehen sich auf den Wirkstoff Natriumvalproat.
Dosierung für Kleinkinder und Kinder: siehe Dosierungstabelle.
Art und Dauer der Anwendung
Die magensaftresistenten Convulex® 150/-300/-500 Kapseln sollten möglichst 1 Stunde vor den Mahlzeiten (morgens nüchtern) unzerkaut mit reichlich Flüssigkeit (z. B. 1 Glas Wasser) eingenommen werden.
Convulex® Tropflösung sollte möglichst zu den Mahlzeiten mit einem halben Glas Wasser oder ähnlichem (ohne Kohlensäure; Hinweis zu Inkompatibilitäten in Abschnitt 6.2 beachten) eingenommen werden.
Die Dauer der Anwendung ist individuell verschieden und wird vom behandelnden Arzt festgelegt.
Die antiepileptische Therapie ist grundsätzlich eine Langzeittherapie.
Über die Einstellung, Behandlungsdauer und das Absetzen von Convulex® sollte im Einzelfall ein Facharzt (Neurologe, Neuropädiater) entscheiden. Im Allgemeinen ist eine Dosisreduktion und ein Absetzen der Medikation frühestens nach zwei- bis dreijähriger Anfallsfreiheit zu erwägen.
Das Absetzen bei anfallsfreien Patienten muss in schrittweiser Dosisreduktion über ein bis zwei Jahre erfolgen. Bei Kindern kann bei der Dosisreduktion das Entwachsen der Dosis pro kg Körpergewicht berücksichtigt werden, wobei sich der EEG-Befund nicht verschlechtern sollte.
4.3 Gegenanzeigen
- Überempfindlichkeit gegen Valproinsäure [Convulex® 150/300/500] bzw. Natriumvalproat [Convulex® Tropflösung] oder einen der sonstigen Bestandteile,
- Lebererkrankungen in der eigenen oder Familienanamnese sowie manifesten schwerwiegenden Leber- und Pankreasfunktionsstörungen,
- Leberfunktionsstörungen mit tödlichem Ausgang während einer Valproinsäure-Therapie bei Geschwistern,
Porphyrie,
Blutgerinnungsstörungen.
4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Convulex® darf nur unter besonderer Vorsicht angewendet werden bei:
- Kleinkindern, bei denen die gleichzeitige Behandlung mit mehreren Antiepileptika erforderlich ist,
- mehrfach behinderten Kindern und Jugendlichen mit schweren Anfallsformen.
Besondere Vorsicht ist auch geboten bei Patienten mit:
- Knochenmarkschädigungen,
- metabolischen Erkrankungen, insbesondere angeborenen Enzymopathien,
- Niereninsuffizienz und Hypoproteinämie,
- systemischem Lupus erythematodes.
Risikogruppen:
siehe Warnhinweise und Sonstige Hinweise Warnhinweise
Über suizidale Gedanken und suizidales Verhalten wurde bei Patienten, die mit Antiepileptika in verschiedenen Indikationen behandelt wurden, berichtet. Eine Metaanalyse randomisierter, placebokontrollierter Studien mit Antiepileptika zeigte auch ein leicht erhöhtes Risiko für das Auftreten von Suizidgedanken und suizidalem Verhalten. Der Mechanismus für die Auslösung dieser Nebenwirkung ist nicht bekannt, und die verfügbaren Daten schließen die Möglichkeit eines erhöhten Risikos bei der Einnahme von Valproinsäure/Natriumvalproat nicht aus.
Deshalb sollten Patienten hinsichtlich Anzeichen von Suizidgedanken und suizidalen Verhaltensweisen überwacht und eine geeignete Behandlung in Erwägung gezogen werden. Patienten (und deren Betreuern) sollte geraten werden, medizinische Hilfe einzuholen, wenn Anzeichen für Suizidgedanken oder suizidales Verhalten auftreten.
Gelegentlich sind schwere Schädigungen der Leber mit tödlichem Ausgang beobachtet worden.
Am häufigsten betroffen sind Säuglinge und Kleinkinder unter 3 Jahren, die an schweren epileptischen Anfällen leiden, besonders wenn zusätzlich eine Hirnschädigung, mentale Retardierung oder eine angeborene Stoffwechselerkrankung vorliegen. Bei dieser Patientengruppe sollte die ValproinsäureAnwendung mit besonderer Vorsicht und als Monotherapie erfolgen. Die Erfahrung hat gezeigt, dass oberhalb dieser Altersgruppe (vor allem jenseits des 10. Lebensjahres) die Häufigkeit der Lebererkrankungen beträchtlich abnimmt.
In der Mehrzahl der Fälle wurden Leberschäden innerhalb der ersten 6 Monate der Therapie beobachtet, insbesondere zwischen der 2. und 12. Woche und zumeist bei der gleichzeitigen Anwendung anderer Antiepileptika.
Bei Patienten mit Leberfunktionsstörungen soll jegliche gleichzeitig Einnahme von Salicylaten gestoppt werden, da diese dem gleichen Metabolismus unterliegen und daher das Risiko von Leberversagen erhöht wird.
Sonstige Hinweise
Bei Patienten mit Niereninsuffizienz und Hypoproteinämie muss der Anstieg an freier Valproinsäure im Serum in Betracht gezogen und die Dosis entsprechend reduziert werden (siehe Abschnitt 5.2).
Die Pharmakokinetik von Valproinsäure kann bei älteren Patienten verändert sein. Die Dosierung sollte anhand der Anfallskontrolle bestimmt werden (siehe Abschnitt 5.2).
Die Anwendung von valproinsäurehaltigen Arzneimitteln führt nur selten zu Reaktionen des Immunsystems. Trotzdem sollte bei Patienten, die Anzeichen eines Lupus erythematodes zeigen, der Einsatz nur unter sorgfältiger Nutzen-Risiko-Abwägung erfolgen.
Die gleichzeitige Anwendung von Valproinsäure/Valproaten und Carbapenemen wird nicht empfohlen (siehe Abschnitt 4.5).
Besondere Vorsichtshinweise für den Gebrauch und Kontrollmaßnahmen
Schwere, lebensbedrohende Schädigungen von Leber oder Pankreas treten gelegentlich auf und kommen fast ausschließlich in den ersten 6 Behandlungsmonaten vor. Betroffen sind vorwiegend Kinder unter 15 Jahren, besonders mehrfachbehinderte Kleinkinder (unter 3 Jahren) und Kinder unter Kombinationstherapie.
Meistens zeigen sich klinische Auffälligkeiten (Appetitverlust, Übelkeit, Erbrechen, Bauchschmerzen, Abneigung gegen gewohnte Speisen, Abneigung gegen Valproinsäure, Müdigkeit, Schlappheit, Zunahme von Frequenz/Schwere der Anfälle, Hämatome/Epistaxis, Ödeme der Augenlider/unteren Extremitäten, Ikterus) schon vor der Veränderung von Laborwerten. Der klinischen Überwachung der Patienten kommt deshalb größere Bedeutung zu als den Laborbefunden. Da es schwierig ist anzugeben, welche Untersuchung, wenn überhaupt, aussagekräftig ist, können Tests, welche die Proteinsynthese widerspiegeln, z.B. die Thromboplastinzeit, die größte Relevanz haben. Schwere Anfälle oder schwere neurologische Störungen in Kombination mit antiepileptischer Behandlung können Risikofaktoren für das Auftreten einer schweren Pankreatitis sein.
Maßnahmen zur Früherkennung einer Leber- und Bauchspeicheldrüsenschädigung:
Vor Behandlungsbeginn sind ausführliche klinische Untersuchungen (insbesondere hinsichtlich Stoffwechselstörungen, Hepatopathie, Pankreasaffektionen und Gerinnungsstörungen) und laborchemische Bestimmung von Blutbild mit Thrombozyten, Bilirubin, SGOT, SGPT, gamma-GT, Lipase, alpha-Amylase im Blut, Blutzucker, Gesamteiweiß, Quick, PTT, Fibrinogen, FibrinogenAbbauprodukten, Faktor VIII und assoziierten Faktoren durchzuführen. Die Patienten sind engmaschig zu überwachen (besonders bei Fieber), die Eltern / Bezugspersonen sind auf mögliche Zeichen einer Leberschädigung (s. o.) hinzuweisen und in die Überwachung mit einzubeziehen.
Eltern und behandelnder Arzt sollten in den ersten 6 Behandlungsmonaten engen direkten oder telefonischen Kontakt halten:
Erster Telefonkontakt 2 Wochen nach Behandlungsbeginn, erste ärztliche und labor-chemische Untersuchung nach 4 Wochen. Danach Arztkontakte jeweils in den Wochen 8, 12, 16, 22, 28, 40 und 52. Telefonkontakte in den Wochen 6, 10, 14, 19, 34.
Eltern sind anzuweisen, bei klinischen Auffälligkeiten und unabhängig von diesem Zeitplan sofort den behandelnden Arzt zu informieren. Laborkontrollen bei den Arztbesuchen:
Bei unauffälligem Kind: Blutbild mit Thrombozyten, SGOT und SGPT, bei jeder zweiten ärztlichen Untersuchung, außerdem Gerinnungsparameter. Nach 12monatiger Therapie ohne Auffälligkeiten sind nur noch 2 - 3 ärztliche Kontrollen pro Jahr erforderlich.
Ein sofortiger Therapieabbruch ist zu erwägen bei:
nicht erklärbarer Störung des Allgemeinbefindens, klinischen Zeichen einer Leber- oder Pankreasaffektion oder Blutungsneigung, mehr als 2 - 3facher Erhöhung der Lebertrans-aminasen auch ohne klinische Zeichen (Enzyminduktion durch evtl. Begleitmedikation bedenken), leichter (eineinhalb- bis zweifacher) Erhöhung der Lebertransaminasen bei gleichzeitigem, akut fieberhaftem Infekt, ausgeprägter Störung des Gerinnungsstatus.
Bei Jugendlichen (etwa ab dem 15. Lebensjahr) und Erwachsenen sind im ersten Halbjahr monatliche Kontrollen des klinischen Befundes und der Laborparameter sowie in jedem Fall vor Therapiebeginn anzuraten.
Weitere Vorsichtshinweise
Unter der Behandlung mit valproinsäurehaltigen Präparaten kann es zu einem Anstieg des Ammoniakserumspiegels (Hyperammonämie) kommen. Deshalb ist beim Auftreten von Symptomen wie Apathie, Somnolenz, Erbrechen, Hypotension sowie bei der Zunahme der Anfallsfrequenz der Serumspiegel von Ammoniak und Valproinsäure zu bestimmen; gegebenenfalls ist die Dosis des Präparates zu reduzieren. Bei Verdacht auf eine bereits bestehende enzymatische Störung des Harnstoffzyklus sollte der Ammoniakserumspiegel bereits vor Beginn der Therapie mit valproinsäurehaltigen Arzneimitteln bestimmt werden.
Zu beachten ist, dass zu Beginn einer Valproinsäure-Behandlung selten auch eine harmlose, meist vorübergehende Übelkeit, manchmal auch mit Erbrechen und Appetitlosigkeit, auftreten kann, die sich von selbst oder bei Dosisverringerung wieder zurückbildet.
Es sollte darauf geachtet werden, dass die Patienten möglichst nicht gleichzeitig saure Getränke oder eisgekühlte Speisen mit Convulex® zu sich nehmen.
Bei der Beobachtung nicht-dosisabhänigiger Nebenwirkungen ist das Absetzen des Arzneimittels angezeigt.
Vor einem operativen Eingriff ist der Gerinnungsstatus zu überprüfen. Bei gleichzeitiger Einnahme von Vitamin-K-Antagonisten wird eine engmaschige Kontrolle des Quick-Wertes empfohlen.
Patienten mit vorausgegangener Knochenmarkschädigung müssen streng überwacht werden.
Valproinsäure führt sehr häufig zu Gewichtszunahme, die ausgeprägt und fortschreitend sein kann. Alle Patienten sind zu Behandlungsbeginn auf dieses Risiko hinzuweisen, und geeignete Maßnahmen sind festzulegen, um die Gewichtszunahme zu minimieren.
Abhängig von der Plasmakonzentration kann Valproinsäure Schilddrüsenhormone aus deren Plasmaproteinbindungen verdrängen und deren Metabolisierung erhöhen, was zur falschen Diagnose einer Schilddrüsenunterfunktion führen kann.
Convulex® 150 / -300 / -500: Patienten mit der seltenen hereditären Fructose-Intoleranz sollten Convulex® 150 / -300 / -500 nicht einnehmen.
4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Bei der Kombination von Convulex® mit anderen Antikonvulsiva ist zu beachten, dass wechselseitige Beeinflussungen der Wirkstoffkonzentrationen im Serum möglich sind.
a) Valproinsäure wird beeinflusst von:
Enzyminduzierende Antiepileptika wie Phenobarbital, Phenytoin, Primidon und Carbamazepin erhöhen die Valproinsäure-Ausscheidung und vermindern dadurch die Wirkung.
Felbamat erhöht dosisabhängig die Serumkonzentrationen von freier Valproinsäure linear um 18 %.
Mefloquin und Meropenem verstärken den Abbau von Valproinsäure und besitzen außerdem potentiell krampfauslösende Wirkungen. Eine gleichzeitige Anwendung kann daher zu epileptischen Anfällen führen.
Chloroquin kann eine Herabsetzung der Krampfschwelle bewirken.
Die Valproinsäurekonzentration im Serum kann durch gleichzeitige Gabe von Cimetidin, Erythromycin und Fluoxetin erhöht werden. Es sind jedoch auch Fälle beschrieben, in denen die Valproinsäurekonzentration im Serum durch gleichzeitige Fluoxetineinnahme erniedrigt wurde.
Ein Absinken der Serumkonzentrationen von Valproinsäure wurde beschrieben, wenn gleichzeitig Carbapeneme angewendet wurden, was zu einer 60-100%igen Senkung der Valproinsäurespiegel in etwa 2 Tagen führte. Aufgrund des raschen Eintritts und des Ausmaßes des Absinkens werden die Folgen einer Wechselwirkung zwischen Valproinsäure und Carbapenemen bei Patienten, die stabil auf Valproinsäure eingestellt sind, als nicht kontrollierbar angesehen und eine gleichzeitige Anwendung sollte daher vermieden werden (siehe Abschnitt 4.4).
Cholestyramin kann die Resorption von Valproinsäure verringern.
Bei gleichzeitiger Einnahme von valproinsäurehaltigen Arzneimitteln und Präparaten mit ausgeprägter Plasmaeiweißbindung, wie z. B. Acetylsalicylsäure, können die Plasmaspiegel der freien Valproinsäure erhöht sein. Eine gleichzeitige Gabe von valproinsäurehaltigen Arzneimitteln und Acetylsalicylsäure sollte bei Kindern unter 12 Jahren unterbleiben und bei Jugendlichen nur nach strenger Nutzen-Risiko-Bewertung erfolgen ).
b) Valproinsäure beeinflusst:
Von besonderer klinischer Bedeutung ist die Erhöhung der Phenobarbital-Konzentration durch Valproinsäure, was sich in einer starken Sedierung (besonders bei Kindern) äußern kann. Falls diese auftritt, muss die Phenobarbital- bzw. Primidondosis erniedrigt werden. (Primidon wird z. T. zu Phenobarbital metabolisiert). Deshalb ist insbesondere innerhalb der ersten 15 Tage einer Kombinationstherapie eine sorgfältige Überwachung empfehlenswert.
Bei bestehender Therapie mit Phenytoin kann durch die zusätzliche Gabe von Convulex® oder einer Dosiserhöhung von Convulex® die Menge des freien Phenytoin ansteigen (Konzentration des nicht eiweißgebundenen, wirksamen Anteils), ohne dass der Serumspiegel des Gesamtphenytoins erhöht ist. Dadurch kann das Risiko für das Auftreten von Nebenwirkungen, insbesondere einer Hirnschädigung, erhöht werden (siehe Abschnitt 4.8).
In der Kombinationstherapie von Valproinsäure mit Carbamazepin wurden Symptome beschrieben, die möglicherweise auf die Potenzierung des toxischen Effektes von Carbamazepin durch Valproinsäure zurückzuführen sind. Klinisches Monitoring ist insbesondere zu Beginn der Kombinationstherapie angezeigt; die Dosis sollte bei Bedarf angepasst werden.
Die gerinnungshemmende Wirkung von Warfarin, anderen Antikoagulantien vom Cumarin-Typ sowie die thrombozytenaggregationshemmende Wirkung von Acetylsalicylsäure können infolge ihrer Verdrängung aus der Plasmaeiweißbindung durch Valproinsäure erhöht sein. Es kann zu erhöhter Blutungsneigung kommen. Die Thromboplastinzeit sollte bei Anwendung von oralen Antikoagulantien engmaschig überwacht werden (siehe Abschnitt 4.4).
Valproat verdrängte bei gesunden Probanden Diazepam aus der Plasmaalbuminbindung und hemmte seinen Metabolismus. In Kombinationsbehandlung kann die Konzentration von ungebundenem Diazepam erhöht sowie die Plasmaclearance und das Verteilungsvolumen der freien DiazepamFraktion (um 25 %; 20 %) reduziert werden. Die Halbwertszeit bleibt jedoch unverändert.
Die gleichzeitige Behandlung mit Valproat und Lorazepam hatte bei Gesunden eine Erniedrigung der Plasmaclearance von Lorazepam um bis zu 40 % zur Folge.
Der Serumspiegel von Phenytoin bei Kindern kann nach gleichzeitiger Verabreichung von Clonazepam und Valproinsäure erhöht werden.
Valproinsäure hemmt den Metabolismus von Lamotrigin, dessen Dosierung daher gegebenenfalls angepasst werden sollte. Es gibt Verdachtsmomente, dass bei einer Kombination von Lamotrigin und valproinsäurehaltigen Arzneimitteln das Risiko von Hautreaktionen erhöht ist, da einzelne Fälle schwerer Hautreaktionen berichtet wurden, die innerhalb von 6 Wochen nach Beginn einer Kombinationstherapie auftraten und sich teilweise nach Absetzen der Medikation oder erst nach entsprechender Behandlung zurückbildeten.
Valproinsäure kann den Serumspiegel von Felbamat um ca. 50 % erhöhen.
Auch der Metabolismus und die Proteinbindung von anderen Wirkstoffen wie Codein werden beeinflusst.
In Kombination mit Barbituraten sowie Neuroleptika und Antidepressiva kann Valproinsäure die zentraldämpfende Wirkung dieser Arzneimittel verstärken. Bei entsprechenden Kombinationen sollten die Patienten sorgfältig beobachtet und die Dosierungen gegebenenfalls angepasst werden.
Da Valproinsäure teilweise zu Ketonkörpern metabolisiert wird, sollte bei Diabetikern mit Verdacht auf Ketoazidose eine mögliche falsch-positive Reaktion eines Tests auf Ketonkörper-Ausscheidung berücksichtigt werden.
Valproinsäure erhöht möglicherweise die Serumkonzentration von Zidovudin, was zu verstärkter Toxizität des Zidovudins führen kann.
Die gleichzeitige Anwendung von Temozolomid und Valproinsäure kann eine geringfügige Reduzierung der Clearance von Temozolomid verursachen, was aber nicht als klinisch relevant erachtet wird.
c) Sonstige:
Die Wirkung von empfängnisverhütenden Hormonpräparaten („Pille“) wird durch Valproinsäure in Monotherapie nicht vermindert, da Valproinsäure keine enzyminduzierende Wirkung besitzt.
Es wird darauf hingewiesen, dass potentiell hepatotoxische Arzneimittel, wie auch Alkohol, die Lebertoxizität von Valproinsäure verstärken können.
Bei gleichzeitiger Behandlung mit valproinsäurehaltigen Arzneimitteln und Clonazepam trat bei Patienten mit Anfällen vom Absence-Typ in der Vorgeschichte ein Absence-Status auf.
Bei einer Patientin mit schizoaffektiver Störung trat bei gleichzeitiger Behandlung mit Valproinsäure, Sertralin (Antidepressivum) und Risperidon (Neuroleptikum) eine Katatonie auf.
Vorsicht ist bei der Anwendung von Valproinsäure in Kombination mit neueren Antiepileptika, deren pharmakodynamische Eigenschaften noch nicht hinreichend bekannt sind, geboten.
4.6 Schwangerschaft und Stillzeit
Dieses Arzneimittel sollte nicht während der Schwangerschaft und von Frauen im gebärfähigen Alter verwendet werden, es sei denn, dies ist eindeutig erforderlich (z. B. in Situationen, in denen andere Behandlungen unwirksam sind oder nicht vertragen werden). Frauen im gebärfähigen Alter müssen während der Behandlung eine wirksame Verhütungsmethode anwenden.
Risiken, die mit der Epilepsie und mit Antiepileptika in Verbindung gebracht werden:
Es ist bekannt, dass bei Kindern von Müttern, die während der Schwangerschaft ein Antiepileptikum eingenommen haben, das Risiko für angeborene Fehlbildungen im Vergleich zu Kindern, die von nicht an Epilepsie erkrankten Frauen geboren wurden, um den Faktor 2 - 3 erhöht ist. Die am häufigsten beobachteten Missbildungen sind Schädigungen des ZNS, Herzmissbildungen und Deformationen des Skeletts, Hypospadie und Lippenspalte. Mentale Retardierung oder eine verzögerte geistige und motorische Entwicklung kann bei diesen Kindern auftreten.
Da die gleichzeitige Anwendung mehrerer Antiepileptika (Polytherapie) während der Schwangerschaft zu einer weiteren Erhöhung des Risikos für angeborene Fehlbildungen führen kann, sollte Convulex® bei Frauen im gebärfähigen Alter und besonders während der Schwangerschaft möglichst als Monotherapie und in der niedrigsten wirksamen Dosis angewendet werden. In keinem Fall sollte eine Behandlung mit Convulex® ohne ärztlichen Rat abgebrochen werden, da unkontrollierte Anfälle sowohl für die Mutter als auch für das ungeborene Kind schwerwiegende Konsequenzen haben können.
Frauen im gebärfähigen Alter sollten daher unbedingt auf die Notwendigkeit von Planung und Überwachung einer eventuellen Schwangerschaft hingewiesen werden. Wenn unter einer Behandlung mit Convulex® eine Schwangerschaft eintritt oder wenn die Behandlung mit Convulex® in der Schwangerschaft erforderlich ist, muss die Notwendigkeit einer Anfallskontrolle sorgfältig gegen das mögliche Risiko dieser Therapie für das ungeborene Kind abgewogen werden.
Risiken, die mit Valproinsäure in Verbindung gebracht werden:
Valproinsäureexposition im ersten und frühen zweiten Trimenon der Schwangerschaft ist ursächlich assoziiert mit einem höheren Risiko für Neuralrohrdefekte (Spina bifida - Inzidenz 1 - 2%, Meningomyelozele u. a.), anderen „midline“-Defekten, wie Hypospadie bei männlichen Kindern, skelettalen Missbildungen und Herzmissbildungen. Diese Missbildungen treten in ähnlicher Häufung auch bei anderen Antiepileptika auf. Bilaterale Aplasie des Radius scheint ein seltener aber spezifischer Effekt von valproinsäurehaltigen Arzneimitteln zu sein.
Gleichzeitig ist die Einnahme von Convulex® in der Schwangerschaft mit einer Zunahme von Anomalien wie facialen Dysmorphien, auch in Verbindung mit mentaler Retardierung, Finger-, Zehen- und Nagelanomalien assoziiert. Zudem gibt es Hinweise darauf, dass es zu einer Verminderung des verbalen IQ (mit oder ohne faciale Dysmorphien) kommen kann.
Bei Frauen im gebärfähigen Alter sollte vor Beginn einer Behandlung auf die Notwendigkeit von Planung und Überwachung einer Schwangerschaft hingewiesen werden. Convulex® passiert die Plazenta und erreicht im fetalen Plasma höhere Konzentrationen als im maternalen.
Falls Convulex® unverzichtbar ist, sollte in der Schwangerschaft, besonders im ersten Trimenon, Convulex® in der niedrigsten anfallskontrollierenden Dosis angewendet werden. Da Fehlbildungen mit großer Wahrscheinlichkeit durch Spitzenkonzentrationen im Plasma ausgelöst werden und die Häufigkeit von Neuralrohrdefekten bei Dosierungen > 100 mg/Tag steigt, sollte bei Kinderwunsch, auf jeden Fall jedoch zwischen dem 20. und 40. Schwangerschaftstag, die Tagesdosis in mehreren kleinen Dosen über den Tag verteilt eingenommen werden. Zusätzlich sollte eine regelmäßige Kontrolle der Plasmakonzentration vorgenommen werden, da offenbar bei gleichbleibender Dosierung die Plasmakonzentrationen im Verlauf der Schwangerschaft erheblichen Veränderungen unterliegen können. Eine Kombination mit anderen Antiepileptika erhöht das Fehlbildungsrisiko. Deshalb sollte Valproinsäure, wenn möglich, als Monotherapie angewendet werden.
Eine frühzeitige Folsäuresubstitution (5 mg/Tag) sollte während der Schwangerschaft, möglichst jedoch bereits bei Planung einer Schwangerschaft durchgeführt werden.
Pränataldiagnostische Maßnahmen zur Früherkennung von Schädigungen (Ultraschall und alphaFetoproteinbestimmung) werden empfohlen.
Es liegen Fallberichte über eine Störung der Blutgerinnung (hämorrhagisches Syndrom) bei Neugeborenen vor, deren Mütter während der Schwangerschaft mit Valproat behandelt worden waren. Dieses Syndrom ist auf eine Hypofibrinogenämie zurückzuführen. Auch von Todesfällen durch völliges Fehlen von Fibrin ist berichtet worden. Die Hypofibrinogenämie tritt möglicherweise gemeinsam mit einem Abfall von Gerinnungsfaktoren auf. Dennoch muss dieses Syndrom von einem Abfall Vitamin-K-abhängiger Gerinnungsfaktoren, der durch Enzyminduktoren wie Phenobarbital verursacht wird, unterschieden werden. Daher sollten Blutplättchen, Fibrinogenspiegel und Gerinnungsfaktoren bei Neugeborenen untersucht und Gerinnungstests durchgeführt werden.
Entzugserscheinungen bei Neugeborenen valproinsäurebehandelter Mütter sind beschrieben worden.
Die Convulex®-Behandlung sollte während der Schwangerschaft nicht ohne ärztliche Zustimmung unterbrochen werden, da ein plötzlicher Therapieabbruch bzw. eine unkontrollierte Verminderung der Dosis zu epileptischen Anfällen der Schwangeren führen kann, die ihr und/oder dem Ungeborenen Schaden zufügen können.
Convulex® tritt in die Muttermilch über. Die Mengen sind jedoch gering und bedeuten im Allgemeinen kein Risiko für das Kind, so dass ein Abstillen in der Regel nicht nötig ist.
4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Zu Beginn einer Therapie mit Convulex®, bei höherer Dosierung oder in Kombination mit am Zentralnervensystem wirkenden Arzneimitteln können zentralnervöse Wirkungen wie z. B. Schläfrigkeit, Verwirrtheit, das Reaktionsvermögen so weit verändern, dass - unabhängig von der Auswirkung des behandelten Grundleidens - die Fähigkeit zur aktiven Teilnahme am Straßenverkehr oder zum Bedienen von Maschinen oder zur Durchführung von Tätigkeiten, die mit Absturz- oder Unfallgefahr einhergehen, beeinträchtigt wird. Dies gilt in verstärktem Maße bei gleichzeitigem Alkoholgenuss.
4.8 Nebenwirkungen
Bei den Häufigkeitsangaben zu Nebenwirkungen werden folgende Kategorien zugrunde gelegt:
Sehr häufig (> 1/10)
Häufig (> 1/100 bis < 1/10)
Gelegentlich (> 1/1.000 bis < 1/100)
Selten (> 1/10.000 bis < 1/1.000)
Sehr selten (< 1/10.000), nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar)
Sehr häufig kann eine isoliert und mäßig ausgeprägte Hyperammonämie ohne Veränderung der Leberfunktionsparameter auftreten, die keinen Therapieabbruch erfordert.
Gelegentlich wurde kurz nach Anwendung von valproinsäurehaltigen Arzneimitteln eine Enzephalopathie beobachtet, deren Pathogenese nicht geklärt ist, und die nach Absetzen des Arzneimittels reversibel ist. Dabei wurden in einigen Fällen erhöhte Ammoniakspiegel sowie bei Kombinationstherapie mit Phenobarbital ein Anstieg des Phenobarbitalspiegels beschrieben. Selten wurden, vor allem bei höherer Dosierung oder in Kombinationstherapie mit anderen Antiepileptika, auch über chronische Enzephalopathien mit neurologischer Symptomatik sowie Störungen höherer kortikaler Funktionen berichtet, deren Pathogenese ebenfalls nicht ausreichend geklärt wurde. Sehr selten wurde über das Auftreten von Koma sowie von Demenz, vergesellschaftet mit zerebraler Atrophie, die nach Absetzen der Medikation reversibel waren berichtet.
Dosisabhängig werden sehr häufig Gewichtszunahme oder häufig Gewichtsabnahme, erhöhter Appetit oder auch Appetitlosigkeit, Schläfrigkeit, Tremor oder Parästhesien beobachtet. Die Gewichtszunahmen können ausgeprägt und fortschreitend verlaufen (siehe Abschnitt 4.4). Ebenfalls dosisabhängig wird häufig vorübergehender Haarausfall beobachtet. Das Haar beginnt üblicherweise innerhalb von 6 Monaten nachzuwachsen, kann dann jedoch gewellter sein als vorher.
Gelegentlich wurden Hypersalivationen, Diarrhoe oder Obstipation, periphere Ödeme, Blutungen, Kopfschmerzen, Spastizität, Ataxie, Schwindel, Reizbarkeit, Hyperaktivität, Aggression, Verwirrtheit, Verhaltensauffälligkeiten, besonders zu Beginn der Behandlung, berichtet.
Ebenfalls gelegentlich wurden Fälle von Stupor beobachtet, die zum Teil mit einer erhöhten Anfallsfrequenz verbunden waren und deren Symptomatik sich bei Reduktion der Dosis oder Absetzen des Arzneimittels zurückbildete. Die Mehrzahl dieser Fälle trat bei einer Kombinationstherapie (insbesondere mit Phenobarbital) oder nach einer raschen Dosiserhöhung auf.
Besonders wurden zu Beginn der Therapie gelegentlich- bei Gabe der Lösung: häufig -gastrointestinale Störungen (Übelkeit - manchmal auch mit Erbrechen und Appetitlosigkeit (siehe Abschnitt 4.4 unter „Weitere Vorsichtshinweise“) -, Magenschmerzen) beobachtet, die sich gewöhnlich trotz Beibehalten der Therapie nach wenigen Tagen zurückbildeten.
Weiterhin wurden Tinnitus, Halluzinationen sowie bei Kindern Enuresis beobachtet.
Von Depressionen wurde berichtet.
Häufig kommt es zu Sedierung, gewöhnlich bei Kombinationsbehandlung mit anderen Antiepileptika. Bei Monotherapie tritt diese Wirkung in seltenen Fällen in einem frühen Behandlungsstadium auf und ist im Allgemeinen vorübergehend.
Selten wurde von Nystagmus berichtet.
Sehr selten wurde über das Auftreten reversibler extrapyramidaler Störungen einschließlich Parkinsonismus berichtet.
Sehr selten wurde über das Auftreten von Hirsutismus und Akne berichtet.
Häufig tritt eine Thrombozytopenie oder Leukopenie auf, die sich oft unter Beibehalten der Medikation, aber immer nach Absetzen von Valproinsäure vollständig zurückbildet. Sehr selten kann eine Beeinträchtigung der Knochenmarksfunktion zu Lymphopenien, Neutropenien, Panzytopenie oder Anämie führen.
Valproinsäure kann zu einer erniedrigten Konzentration von Fibrinogen bzw. Faktor VIII führen sowie die sekundäre Phase der Plättchenaggregation hemmen und dadurch eine verlängerte Blutungszeit bedingen.
Die Einnahme von Convulex® führte selten zu Reaktionen der Haut (Erythema multiforme). Allergische Reaktionen (von Ausschlägen bis hin zu Überempfindlichkeitsreaktionen) wurden beobachtet. Selten wurden auch Veränderungen in den immunologischen Abwehrmechanismen (Lupus erythematodes) beobachtet. Gelegentlich wurde über das Auftreten von Vaskulitis berichtet. Daneben wurden einzelne Ausnahmefälle von schweren Hautreaktionen (Stevens-Johnson-Syndrom und toxische epidermale Nekrolyse bzw. Lyell-Syndrom) berichtet.
Selten wurde über erhöhte Testosteronspiegel, polyzystische Ovarien, Dysmenorrhoe und Amenorrhoe berichtet. Gynäkomastie trat sehr selten auf.
Selten wurde nach Absetzen von valproinsäurehaltigen Arzneimitteln vom Auftreten eines reversiblen Fanconi-Syndroms (metabolische Acidose, Phosphaturie, Aminoacidurie, Glucosurie) in der Literatur berichtet.
Bei einer Langzeittherapie mit Convulex®, zusammen mit anderen Antiepileptika, insbesondere Phenytoin, kann es zu Zeichen einer Hirnschädigung (Enzephalopathie) kommen: vermehrte Krampfanfalle, Antriebslosigkeit, Stupor, Muskelschwäche (muskuläre Hypotonie), Bewegungsstörungen (Choreatiforme Dyskinesien) und schwere Allgemeinveränderungen im EEG.
Gelegentlich kommen dosisunabhängig auftretende schwerwiegende (bis tödlich verlaufende) Leberfunktionsstörungen vor. Bei Kindern, besonders in der Kombinationstherapie mit anderen Antiepileptika, ist das Risiko der Leberschädigung deutlich erhöht (siehe Abschnitt 4.4).
Selten wurde auch von Porphyrie berichtet.
Selten ist über eine Schädigung der Bauchspeicheldrüse, teilweise mit tödlichem Ausgang, berichtet worden.
Über reversiblen oder irreversiblen Hörverlust wurde berichtet, wobei ein kausaler Zusammenhang mit valproinsäurehaltigen Arzneimitteln jedoch nicht gesichert ist.
Besondere Aufmerksamkeit muss im Laufe der Behandlung auf folgende Anzeichen einer Leberschädigung gerichtet werden:
Verringerung antiepileptischer Wirkung, die durch erneutes Auftreten oder Zunahme epileptischer Anfälle gekennzeichnet ist; länger andauernde Symptome wie körperliches Schwächegefühl, Teilnahmslosigkeit, Appetitlosigkeit, Übelkeit und wiederholtes Erbrechen oder unklare Oberbauchbeschwerden, vermehrte Gewebewassereinlagerungen im ganzen Körper oder in Teilen davon, Bewusstseinsstörungen mit Verwirrtheit, Unruhe oder Bewegungsstörungen.
Selten wurden auch Schädigungen der Bauchspeicheldrüse mit ähnlichen Beschwerden beobachtet. Hinsichtlich dieser Anzeichen sollten Säuglinge und Kleinkinder ärztlich engmaschig überwacht werden.
Sind die oben erwähnten Beschwerden anhaltend oder schwerwiegend, so sind neben einer gründlichen Untersuchung auch entsprechende Laboruntersuchungen vorzunehmen (siehe Abschnitt 4.4).
Es gibt Fallberichte über die Abnahme der Knochendichte unter dem Bild der Osteoporose bis hin zu pathologischen Frakturen bei Patienten, die Valproinsäure/Natriumvalproat über eine lange Zeit angewendet haben. Der Mechanismus, über den Valproinsäure/Natriumvalproat den KnochenMetabolismus beeinflusst, ist nicht bekannt.
4.9 Überdosierung
Bei jeder Beurteilung einer Intoxikation sollte an die Möglichkeit einer Mehrfach-Intoxikation z. B. durch Einnahme mehrerer Arzneimittel, beispielsweise in suizidaler Absicht, gedacht werden.
Valproinsäure besitzt bei therapeutischen Serumspiegeln (Bereich 50 - 100 ^g/ml) eine relativ geringe Toxizität. Sehr selten sind akute Intoxikationen mit Valproinsäure bei Serumspiegeln über 100 ^g/ml bei Erwachsenen und auch bei Kindern vorgekommen.
Einzelfälle akuter und chronischer Überdosierungen mit tödlichem Ausgang sind aus der Literatur bekannt.
Symptome einer Überdosierung:
Das Vergiftungsbild ist gekennzeichnet durch Verwirrtheitszustände, Sedation bis hin zum Koma, Muskelschwäche und Hypo- bzw. Areflexie.
In Einzelfällen wurden Hypotension, Miosis, kardiovaskuläre wie respiratorische Störungen, zerebrales Ödem, intrakranielle Drucksteigerung, metabolische Azidose, Hypernatriämie beobachtet. Hohe Serumspiegel riefen bei Erwachsenen wie bei Kindern abnorme neurologische Störungen wie z. B. erhöhte Anfallsneigung und Verhaltensänderungen hervor.
Maßnahmen bei Überdosierung:
Ein spezifisches Antidot ist nicht bekannt.
Die Therapie muss sich deshalb auf allgemeine Maßnahmen zur Entfernung des Wirkstoffes aus dem Organismus und Stützung der Vitalfunktionen beschränken.
Wenn möglich ist initial, innerhalb von 30 Minuten nach Einnahme, Erbrechen auszulösen bzw. Magenspülung und die Gabe von Aktivkohle vorzunehmen. Hierbei ist intensivmedizinische Überwachung erforderlich.
Hämodialyse und forcierte Diurese können wirksam sein. Die Peritonealdialyse ist wenig wirksam. Über die Wirksamkeit der hämatogenen Kohleperfusion sowie der kompletten Plasmasubstitution und -transfusion liegen keine ausreichenden Erfahrungen vor. Aus diesem Grund wird eine intensive internistische Therapie ohne spezielle Detoxikationsverfahren, besonders bei Kindern, aber mit Kontrolle der Serumkonzentration empfohlen.
Die intravenöse Gabe von Naloxon zur Aufhellung der Bewusstseinstrübung ist in einem Fall als wirksam beschrieben worden.
5. PHARMAKOLOGISCHE EIGENSCHAFTEN
5.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Antiepileptika, Fettsäurederivate ATC-Code: N03AG01
Valproinsäure ist ein Antiepileptikum, das keine strukturelle Ähnlichkeit mit anderen antikonvulsiven Wirkstoffen zeigt. Als Wirkmechanismen von Valproinsäure werden eine Erhöhung der GABA-mediierten Inhibition durch einen präsynaptischen Effekt auf den GABA-Metabolismus und/oder eine direkte postsynaptische Wirkung auf die Ionenkanälchen der neuronalen Membran angenommen. Valproinsäure ist in Wasser sehr schwer löslich (1:800), das Natriumsalz ist in Wasser sehr leicht löslich (1:0,4).
5.2 Pharmakokinetische Eigenschaften
- Resorption
Nach oraler Gabe werden die Valproinsäure und ihr Natriumsalz im Gastrointestinaltrakt schnell und nahezu vollständig resorbiert.
- Serumspiegel, Plasmaproteinbindung, Verteilung
Der Zeitpunkt der maximalen Serumkonzentration hängt von der galenischen Darreichungsform ab:
Bei Lösungen wird sie innerhalb von 0,5 - 2 Stunden erreicht.
Bei magensaftresistenten Zubereitungen ergeben sich maximale Serumkonzentrationen nach 2 - 8 Stunden mit einer Verzögerung von 1 - 4 Stunden. Hierbei wurden nach einer Dosis von 600 mg maximale Serumkonzentrationen zwischen 46 - 88 ^g/ml gemessen.
Es besteht keine lineare Beziehung zwischen Dosis und Serumkonzentration.
Der mittlere therapeutische Bereich der Serumkonzentration wird mit 50 - 100 ^g/ml angegeben. Oberhalb von 100 ^g/ml ist vermehrt mit Nebenwirkungen bis hin zur Intoxikation zu rechnen. Steady-State-Serumspiegel werden in der Regel innerhalb von 2 Wochen erreicht.
In der Zerebrospinalflüssigkeit liegen die Valproinsäure-Konzentrationen bei 10 % der jeweiligen Serumkonzentration.
Das V erteilungsvolumen ist altersabhängig und beträgt in der Regel 0,13 - 0,23 l/kg, bei Jüngeren 0,13 -0,19 l/kg.
Valproinsäure wird zu 90 - 95 % an Plasmaproteine gebunden, vornehmlich an Albumin. Bei höherer Dosierung nimmt die Eiweißbindung ab.
Ältere Patienten:
Die Pharmakokinetik von Valproinsäure kann bei älteren Patienten wegen eines erhöhten Verteilungsvolumens und einer verminderten Proteinbindung, die zu einer Erhöhung der freien Wirkstoffkonzentration führen kann, verändert sein.
Patienten mit Niereninsuffizienz:
Die Pharmakokinetik der Valproinsäure kann bei Patienten mit renaler Insuffizienz aufgrund der verminderten Proteinbindung, die zu einer Erhöhung der freien Wirkstoffkonzentration führt, verändert sein. In einer Studie wurden erhöhte Werte freien Wirkstoffes (8,5 bis über 20 %) bei Patienten mit signifikant verminderter Nierenfunktion beobachtet.
Die Valproinsäuregesamtkonzentration, bestehend aus freiem und proteingebundenem Anteil, kann bei Vorliegen einer Hypoproteinämie jedoch im wesentlichen unverändert sein, sie kann aber auch aufgrund der vermehrten Metabolisierung des freien Anteils vermindert sein.
- Metabolismus, Ausscheidung
Die Biotransformation erfolgt über Glukuronidierung sowie ß-, ©- und ©-1-Oxidation. Etwa 20 % der applizierten Dosis treten nach renaler Exkretion als Ester-Glukuronid im Harn auf. Es existieren mehr als 20 Metabolite, wobei die der ©-Oxidation als hepatotoxisch angesehen werden. Weniger als 5 % der applizierten Dosis Valproinsäure erscheinen unver-ändert im Urin.
Hauptmetabolit ist die 3-Keto-Valproinsäure, die zu 3 - 60 % im Harn auftritt. Dieser Metabolit ist bei der Maus antikonvulsiv wirksam, beim Menschen ist die Wirkung noch nicht geklärt.
- Plasmaclearance, Plasmahalbwertszeit
Die Plasmaclearance betrug in einer Studie 12,7 ml/min bei Patienten mit Epilepsie, bei Gesunden liegt sie bei 5 - 10 ml/min, bei Einnahme enzyminduzierender Antiepileptika erhöht sie sich.
Die Plasmahalbwertszeit liegt bei Monotherapie durchschnittlich bei 12 - 16 Stunden und bleibt auch bei Langzeittherapie konstant.
Bei Kombination mit anderen Arzneimitteln (z. B. Primidon, Phenytoin, Phenobarbital und Carbamazepin) sinkt die Halbwertszeit auf Werte zwischen 4 und 9 Stunden, in Abhängigkeit von der Enzyminduktion. Neugeborene und Kinder bis zu 18 Monaten zeigen Plasmahalbwertszeiten zwischen 10 und 67 Stunden. Die längsten Halbwertszeiten wurden unmittelbar nach der Geburt beobachtet, oberhalb von 2 Monaten nähern sich die Werte denen von Erwachsenen.
Bei Leberkranken ist die Halbwertszeit verlängert. Im Falle von Überdosierung wurden Halbwertszeiten bis zu 30 Stunden beobachtet.
In der Schwangerschaft nimmt bei Zunahme des Verteilungsvolumens im dritten Trimenon die hepatische und renale Clearance zu, mit einem möglichen Abfall der Serumkonzentration bei gleich hoher Dosierung.
Ferner ist zu beachten, dass im Verlauf der Schwangerschaft sich die Plasmaproteinbindung verändern und der freie (therapeutisch wirkende) Anteil der Valproinsäure zunehmen kann.
- Übergang in die Muttermilch
Valproinsäure ist plazentagängig und geht in die Muttermilch über. Im Steady-State beträgt die Konzentration in der Muttermilch bis ca. 10 % der Serumkonzentration.
Bioverfügbarkeit / Bioäquivalenz
Convulex® 150:
Eine im Jahr 1983 durchgeführte Bioverfügbarkeitsuntersuchung an 8 gesunden Probanden (21 - 58 Jahre, m) ergab nach einmaliger Einnahme von 2 magensaftresistenten Kapseln (bzw. 300 mg Valproinsäure) Convulex® 150 im Vergleich zum Referenzpräparat folgende Werte:
|
Testpräparat |
Referenzpräparat | |
|
maximale Plasmakonzentration (Cmax): |
25,8 ± 7,2 mg/l |
26,6 ± 4,8 mg/l |
|
Zeitpunkt der maximalen Plasmakonzentration (tmax): |
4,0 ± 1,1 h |
3,3 ± 1,3 h |
|
Fläche unter der KonzentrationsZeitkurve (AUC): |
313 ± 73 mg/l x h |
351 ± 59 mg/l x h |
Angabe der Werte als Mittelwert und Streubreite
Mittlere Plasmaspiegelverläufe im Vergleich zu einem Referenzpräparat in einem Konzentrations-ZeitDiagramm:
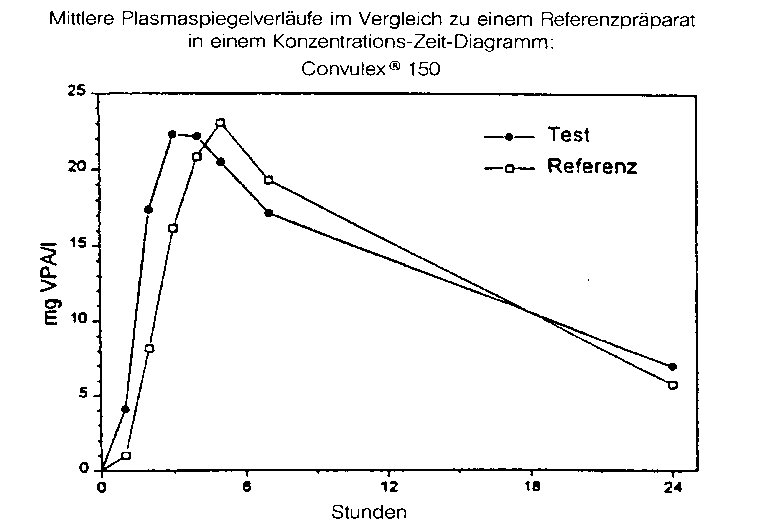
|
Testpräparat |
Referenzpräparat | |
|
maximale Plasmakonzentration (Cmax): |
30,0 ± 4,1 mg/l |
26,7 ± 4,5 mg/l |
|
Zeitpunkt der maximalen Plasmakonzentration (tmax): |
2,8 ± 1,0 h |
2,8 ± 0,8 h |
|
Fläche unter der KonzentrationsZeitkurve (AUC): |
355 ± 49 mg/l x h |
317 ± 72 mg/l x h |
Angabe der Werte als Mittelwert und Streubreite
Mittlere Plasmaspiegelverläufe im Vergleich zu einem Referenzpräparat in einem Konzentrations-ZeitDiagramm:
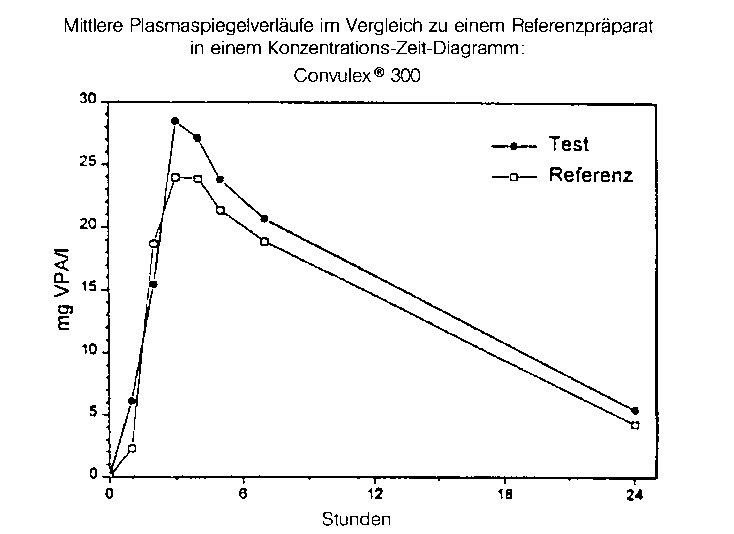
|
Testpräparat |
Referenzpräparat | |
|
maximale Plasmakonzentration (Cmax): |
79,0 ± 22,9 mg/l |
58,5 ± 26,3 mg/l |
|
Zeitpunkt der maximalen Plasmakonzentration (tmax): |
3,3 ± 1,0 h |
4,2 ± 2,1 h |
|
Fläche unter der KonzentrationsZeitkurve (AUC): |
963 ± 358 mg/l x h |
887 ± 399 mg/l x h |
Angabe der Werte als Mittelwert und Streubreite
Mittlere Plasmaspiegelverläufe im Vergleich zu einem Referenzpräparat in einem Konzentrations-ZeitDiagramm:
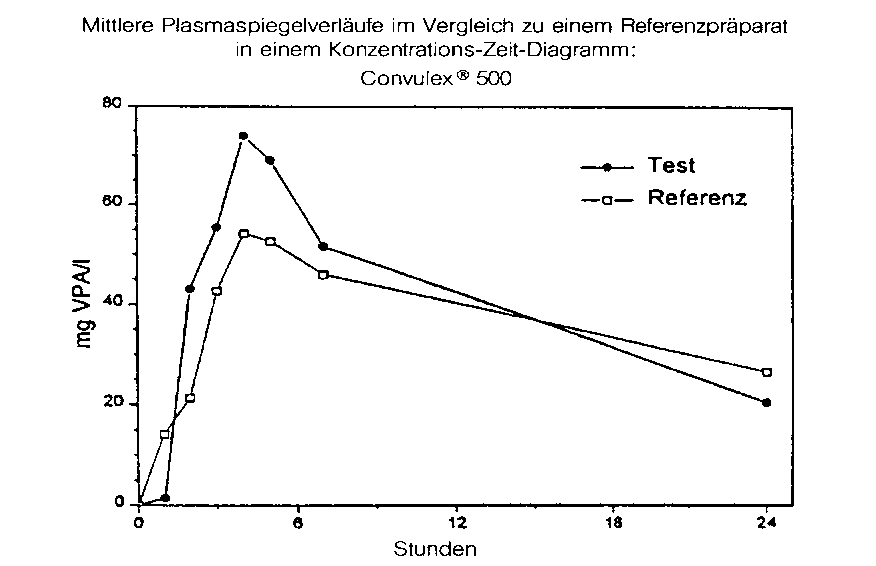
|
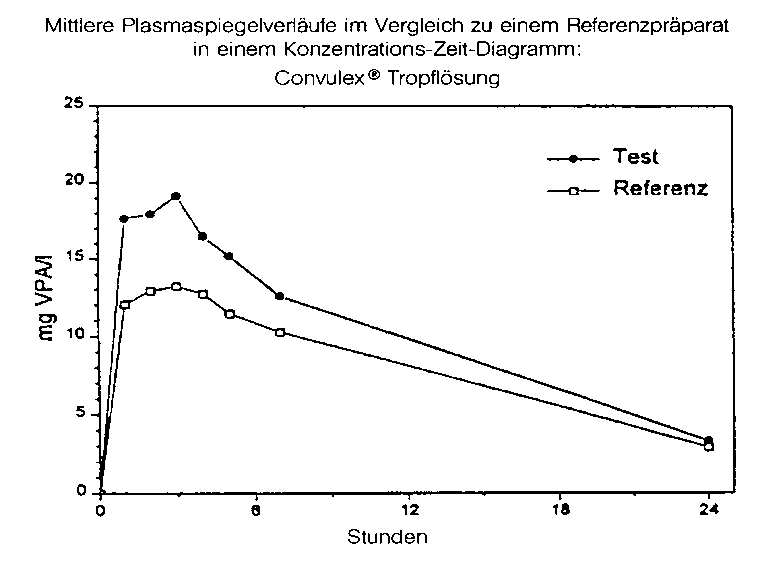
Testpräparat |
Referenzpräparat | |
|
maximale Plasmakonzentration (Cmax): |
21,0 ± 4,3 mg/l |
15,2 ± 3,4 mg/l |
|
Zeitpunkt der maximalen Plasmakonzentration (tmax): |
2,4 ± 1,1 h |
2,3 ± 1,4 h |
|
Fläche unter der KonzentrationsZeitkurve (AUC): |
241 ± 58 mg/l x h |
189 ± 58 mg/l x h |
Angabe der Werte als Mittelwert und Streubreite
Mittlere Plasmaspiegelverläufe im Vergleich zu einem Referenzpräparat in einem Konzentrations-ZeitDiagramm:
5.3 Präklinische Daten zur Sicherheit
a) Akute Toxizität
Untersuchungen zur akuten Toxizität von Natriumvalproat an verschiedenen Tierarten haben LD50-Werte zwischen 1200 und 1600 mg/kg/KG nach oraler Gabe und zwischen 750 und 950 mg/kg/KG nach i.v. Gabe ergeben.
b) Chronische Toxizität
In Untersuchungen zur chronischen Toxizität wurden nach hohen Dosen (250 mg/kg bei Ratten; 90 mg/kg bei Hunden) Atrophie der Hoden, Degeneration des Ductus deferens und eine insuffiziente Spermatogenese sowie Lungen- und Prostataveränderungen festgestellt.
c) Mutagenes und tumorerzeugendes Potential
Mutagenitätstest an Bakterien sowie an Ratten und Mäusen verliefen negativ. Langzeituntersuchungen wurden an Ratten und Mäusen durchgeführt. Bei sehr hohen Dosierungen wurden vermehrt subkutane Fibrosarkome bei männlichen Ratten beobachtet. Valproinsäure erwies sich in Tierstudien als teratogen.
6. PHARMAZEUTISCHE ANGABEN
6.1 Liste der sonstigen Bestandteile
Convulex® 150 / -300 / -500:
Gelatine
Glycerol
Sorbitol (E 420) und andere Polyole Salzsäure 25 %
Titandioxid (E 171)
Methacrylsäure-Ethylacrylat-Copolymer (1:1), Dispersion 30%
Triethylcitrat Macrogol 6000 Glycerolmonostearat Eisenoxid rot (E 172)
Eisenoxid schwarz (E 172)
Schellack
Convulex® Tropflösung:
Saccharin-Natrium Orangenaroma Natriumhydroxid Salzsäure (35 - 39 %) gereinigtes Wasser
6.2 Inkompatibilitäten
Convulex® Tropflösung:
Es wird empfohlen, keine kohlensäurehaltigen Getränke wie Mineralwasser oder ähnliches zum Einnehmen zu verwenden.
6.3 Dauer der Haltbarkeit
5 Jahre
Convulex® Tropflösung ist nach Anbruch des Behältnisses innerhalb von 12 Wochen aufzubrauchen.
6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Convulex® 150, -300 und -500:
In der Originalverpackung aufbewahren, um den Inhalt vor Feuchtigkeit zu schützen.
Nicht über 25°C und nicht im Kühlschrank lagern!
Convulex® Tropflösung:
Die Flasche im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Nicht über 25°C lagern.
6.5 Art und Inhalt des Behältnisses
Convulex® Tropflösung:
Braune Glasflasche mit Sicherheits-Schraubverschluss, in einem Umkarton verpackt, mit beigefügter graduierter Dosierspritze für Zubereitungen zum Einnehmen (Angaben in mg und ml) aus Polyethylen mit einem Polystyrolkolben.
Es liegt folgende Packungsgröße vor:
100 ml Lösung zum Einnehmen (N2)
Convulex® 150:
Al/PVC-PVDC bzw. alternativ Al/PVC Blisterpackungen, in einem Umkarton verpackt.
Es liegen folgende Packungsgrößen vor:
50 magensaftresistente Kapseln (N1)
100 magensaftresistente Kapseln (N2)
Convulex® 300:
Al/PVC-PVDC bzw. alternativ Al/PVC Blisterpackungen, in einem Umkarton verpackt.
Es liegen folgende Packungsgrößen vor:
50 magensaftresistente Kapseln (N1)
100 magensaftresistente Kapseln (N2)
200 magensaftresistente Kapseln (N3)
1000 (10 x 100) magensaftresistente Kapseln (Klinikpackung)
Convulex® 500:
Al/PVC-PVDC bzw. alternativ Al/PVC Blisterpackungen, in einem Umkarton verpackt.
Es liegen folgende Packungsgrößen vor:
50 magensaftresistente Kapseln (N1)
100 magensaftresistente Kapseln (N2)
200 magensaftresistente Kapseln (N3)
1000 (10 x 100) magensaftresistente Kapseln (Klinikpackung)
6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung
Keine besonderen Anforderungen.
7. INHABER DER ZULASSUNG
G.L. Pharma GmbH Schlossplatz 1 A - 8502 Lannach Österreich
8. ZULASSUNGSNUMMERN
Convulex® 150: 2241.01.00
|
Convulex® 300: Convulex® 500: Convulex® Tropflösung: |
2241.02.00 2241.00. 00 2241.00. 01 |
9. DATUM DER ERTEILUNG DER ZULASSUNG / VERLÄNGERUNG DER ZULASSUNG
|
Convulex® 150: Convulex® 300: Convulex® 500: Convulex® Tropflösung: |
03.06.1985 / 17.05.2002 03.06.1985 / 15.05.2002 10.05.1982 / 31.05.2002 03.06.1985 / 17.05.2002 |
10. STAND DER INFORMATION
06/2014
11. VERKAUFSABGRENZUNG
V erschreibungspflichtig
22/22