Drosfemine 0,02 Mg / 3 Mg Filmtabletten
FACHINFORMATION
1. BEZEICHNUNG DES ARZNEIMITTELS
Drosfemine 0,02 mg / 3 mg Filmtabletten
2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
24 rosafarbene Filmtabletten:
Jede Filmtablette enthält 0,02 mg Ethinylestradiol und 3 mg Drospirenon. Sonstiger Bestandteil mit bekannter Wirkung: 46,18 mg Lactose-Monohydrat.
4 weiße (inaktive) Placebo-Filmtabletten:
Die Tablette enthält keine Wirkstoffe.
Sonstiger Bestandteil mit bekannter Wirkung: 50,56 mg Lactose-Monohydrat. Vollständige Auflistung der sonstigen Bestandteile siehe Abschnitt 6.1.
3. DARREICHUNGSFORM
Filmtablette
Die wirkstoffhaltige Tablette ist eine rosafarbene und runde Filmtablette. Die Placebotablette ist eine weiße, runde Filmtablette mit Prägung.
4. KLINISCHE ANGABEN
4.1 Anwendungsgebiete
Orale Kontrazeption.
Bei der Entscheidung, Drosfemine zu verschreiben, sollten die aktuellen, individuellen Risikofaktoren der einzelnen Frauen, insbesondere im Hinblick auf venöse Thromboembolien (VTE), berücksichtigt werden. Auch sollte das Risiko für eine VTE bei Anwendung von Drosfemine mit dem anderer kombinierter hormonaler Kontrazeptiva (KHK) verglichen werden (siehe Abschnitte 4.3 und 4.4).
4.2 Dosierung und Art der Anwendung
Art der Anwendung: Zum Einnehmen.
Wie ist Drosfemine einzunehmen?
Die Tabletten müssen jeden Tag etwa zur gleichen Zeit, falls erforderlich mit etwas Flüssigkeit, in der auf der Blisterpackung angegebenen Reihenfolge eingenommen werden.
Die Tabletteneinnahme erfolgt durchgehend. Täglich muss 1 Tablette über 28 aufeinander folgende Tage eingenommen werden. Mit der jeweils nächsten Packung wird am Tag nach Einnahme der letzten Tablette aus der vorherigen Packung begonnen. Eine Abbruchblutung setzt üblicherweise 2-3 Tage nach Einnahmebeginn der Placebotabletten ein und kann noch andauern, wenn mit der Einnahme aus der nächsten Packung begonnen wird.
Beginn der Einnahme von Drosfemine
Keine vorangegangene Einnahme von hormonalen Kontrazeptiva (im letzten Monat)
Mit der Tabletteneinnahme ist am 1. Tag des natürlichen Zyklus (d. h. am ersten Tag der Menstruationsblutung) zu beginnen.
Wechsel von einem kombinierten hormonalen Kontrazeptivum (kombiniertes orales Kontrazeptivum IKOKI. Vaginalring oder transdermales Pflaster)
Mit der Einnahme von Drosfemine sollte vorzugsweise am Tag nach Einnahme der letzten wirkstoffhaltigen Tablette (die letzte Tablette. die Wirkstoffe enthält) des zuvor eingenommenen KOKs begonnen werden. spätestens aber am Tag nach dem üblichen einnahmefreien oder PlacebotablettenIntervall des zuvor eingenommenen KOKs. Für den Fall. dass ein Vaginalring oder ein transdermales Pflaster angewendet wurde. sollte die Einnahme von Drosfemine vorzugsweise am Tag der Entfernung begonnen werden. spätestens aber. wenn die nächste Applikation fällig wäre.
Wechsel von einem Gestagenmonopräparat („Minipille“. Injektionspräparat, Implantat) oder von einem gestagenfreisetzenden Intrauterinpessar (IUP)
Bei vorheriger Einnahme der Minipille kann an jedem beliebigen Tag gewechselt werden (die Umstellung von einem Implantat oder einem IUP muss am Tag der Entfernung erfolgen. die Umstellung von einem Injektionspräparat zu dem Zeitpunkt. an dem die nächste Injektion fällig wäre). In jedem Fall ist während der ersten 7 Tage der Tabletteneinnahme zusätzlich die Anwendung einer nicht hormonalen Verhütungsmethode (Barrieremethode) erforderlich.
Nach einem Abort im ersten Trimenon
Es kann sofort mit der Einnahme von Drosfemine begonnen werden. In diesem Fall sind keine zusätzlichen empfängnisverhütenden Maßnahmen erforderlich.
Nach einer Geburt oder einem Abort im zweiten Trimenon
Den Anwenderinnen sollte empfohlen werden. an den Tagen 21 bis 28 nach einer Geburt oder nach einem Abort im zweiten Trimenon mit der Einnahme zu beginnen. Bei einem späteren Einnahmebeginn sollte während der ersten 7 Tage zusätzlich eine Barrieremethode angewendet werden. Wenn jedoch bereits Geschlechtsverkehr stattgefunden hat. muss vor Beginn der KOK-Einnahme eine Schwangerschaft ausgeschlossen oder die erste Menstruationsblutung abgewartet werden.
Zur Anwendung bei stillenden Frauen siehe Abschnitt 4.6.
Vorgehen bei vergessener Tabletteneinnahme
Die vergessene Einnahme von Placebotabletten kann vernachlässigt werden. Die vergessenen Placebotabletten sollten jedoch verworfen werden. um eine versehentliche Verlängerung der Placebotablettenphase zu vermeiden. Die folgenden Hinweise beziehen sich nur auf vergessene wirkstoffhaltige Tabletten:
Wird innerhalb von 24 Stunden nach dem üblichen Einnahmezeitpunkt bemerkt. dass die Einnahme einer Tablette vergessen wurde. ist der kontrazeptive Schutz nicht eingeschränkt. Die Frau sollte die Tablette sofort und die darauffolgenden Tabletten wieder zur gewohnten Zeit einnehmen.
Wenn die Einnahme der letzten Tablette mehr als 24 Stunden zurückliegt. ist der Empfängnisschutz nicht mehr voll gewährleistet. Für das Vorgehen bei vergessener Einnahme gelten die folgenden zwei Grundregeln:
1. Das empfohlene hormonfreie Intervall beträgt 4 Tage; die Tabletteneinnahme darf nie länger als 7
Tage unterbrochen werden.
2. Eine regelmäßige Einnahme der Tabletten über mindestens 7 Tage ist erforderlich, um wirkungsvoll die Hypothalamus-Hypophysen-Ovar-Achse zu unterdrücken.
Entsprechend können für die tägliche Praxis folgende Empfehlungen gegeben werden:
Tag 1-7
Die Einnahme der letzten vergessenen Tablette sollte so schnell wie möglich nachgeholt werden, auch wenn dies bedeutet, dass zwei Tabletten zur gleichen Zeit eingenommen werden. Die weitere Tabletteneinnahme erfolgt dann zur gewohnten Zeit. Während der nächsten 7 Tage sollte jedoch zusätzlich eine Barrieremethode, z. B. ein Kondom, angewendet werden. Wenn in den vergangenen 7 Tagen Geschlechtsverkehr stattgefunden hat, sollte die Möglichkeit einer Schwangerschaft in Betracht gezogen werden. Das Risiko einer Schwangerschaft ist umso höher, je mehr Tabletten vergessen wurden und je näher dies zeitlich am regulären Placebo-Intervall liegt.
Tag 8-14
Die Einnahme der letzten vergessenen Tablette sollte so schnell wie möglich nachgeholt werden, auch wenn dies bedeutet, dass zwei Tabletten zur gleichen Zeit eingenommen werden. Die weitere Einnahme der Tabletten erfolgt dann zur üblichen Zeit. Vorausgesetzt, dass die Einnahme der Tabletten an den 7 Tagen vor der ersten vergessenen Tablette korrekt erfolgt ist, besteht keine Notwendigkeit, zusätzliche kontrazeptive Schutzmaßnahmen anzuwenden. Wurde jedoch mehr als 1 Tablette vergessen, sollte die Anwendung zusätzlicher Schutzmaßnahmen über 7 Tage empfohlen werden.
Tag 15-24
Aufgrund der bevorstehenden Placebotablettenphase kann ein voller Empfängnisschutz nicht mehr gewährleistet werden.
Durch eine Anpassung des Einnahmeschemas lässt sich eine Herabsetzung der empfängnisverhütenden Wirkung dennoch verhindern. Bei Einhalten einer der beiden folgenden Vorgehensweisen besteht daher keine Notwendigkeit zusätzlicher kontrazeptiver Maßnahmen, vorausgesetzt, die Tabletteneinnahme an den 7 Tagen vor der ersten vergessenen Tablette erfolgte korrekt. Wenn dies nicht der Fall ist, sollte die Anwenderin wie unter Punkt 1 beschrieben vorgehen und außerdem in den nächsten 7 Tagen zusätzliche Schutzmaßnahmen anwenden.
1. Die Anwenderin sollte die Einnahme der letzten vergessenen Tablette so schnell wie möglich nachholen, auch wenn dies bedeutet, dass zwei Tabletten zur gleichen Zeit eingenommen werden. Die Einnahme der Tabletten erfolgt dann wieder zur üblichen Zeit bis die wirkstoffhaltigen Tabletten aufgebraucht sind. Die 4 Placebotabletten müssen verworfen werden. Mit der Einnahme aus der nächsten Blisterpackung muss sofort begonnen werden. Es ist unwahrscheinlich, dass es bei der Anwenderin vor Aufbrauchen der wirkstoffhaltigen Tabletten aus der zweiten Packung zu einer Abbruchblutung kommt, allerdings können noch während der Einnahme Schmier- oder Durchbruchblutungen auftreten.
2. Es kann auch ein Abbruch der Einnahme der wirkstoffhaltigen Tabletten aus der aktuellen Blisterpackung empfohlen werden. In diesem Fall sollten bis zu 4 Tage lang Placebotabletten eingenommen werden, die Tage der vergessenen Tabletteneinnahme eingerechnet. Danach wird mit der Tabletteneinnahme aus der neuen Packung begonnen.
Bei vergessener Tabletteneinnahme und anschließendem Ausbleiben einer Abbruchblutung in der Placebotablettenphase sollte die Möglichkeit einer Schwangerschaft in Betracht gezogen werden.
Verhalten bei gastrointestinalen Störungen
Bei schweren gastrointestinalen Störungen (z. B. Erbrechen oder Durchfall) werden die Wirkstoffe möglicherweise nicht vollständig aufgenommen und zusätzliche kontrazeptive Maßnahmen sind erforderlich. Bei Erbrechen in den ersten 3 - 4 Stunden nach der Tabletteneinnahme sollte so schnell wie möglich eine weitere (Ersatz-)Tablette eingenommen werden.
Die Einnahme der neuen Tablette sollte, wenn möglich, innerhalb von 24 Stunden nach der normalen Einnahmezeit erfolgen.
Wenn mehr als 24 Stunden vergangen sind, empfiehlt sich die im Abschnitt 4.2 unter „Vorgehen bei vergessener Tabletteneinnahme“ genannte Vorgehensweise für vergessene Tabletten. Wenn die Anwenderin nicht von ihrem normalen Einnahmerhythmus abweichen möchte, muss sie die Ersatztablette(n) aus einer anderen Blisterpackung einnehmen.
Verschiebung oder Verzögerung der Menstruation
Um die Entzugsblutung hinauszuschieben, soll direkt mit der Einnahme aus der nächsten Blisterpackung Drosfemine begonnen werden, ohne die Placebotabletten aus der aktuellen Packung einzunehmen. Die Einnahme kann so lange fortgesetzt werden wie gewünscht, maximal bis die wirkstoffhaltigen Tabletten der zweiten Packung aufgebraucht sind. Während der Einnahme aus der zweiten Packung kann es zu Durchbruch- oder Schmierblutungen kommen. Nach der Placebotablettenphase kann die Einnahme von Drosfemine wie üblich fortgesetzt werden.
Zum Verschieben der Entzugsblutung auf einen anderen Wochentag als nach dem bisherigen Einnahmeschema üblich, kann die bevorstehende Placebotablettenphase um die gewünschte Anzahl von Tagen verkürzt werden. Je kürzer das Intervall, desto höher ist die Wahrscheinlichkeit einer ausbleibenden Entzugsblutung und während der Einnahme aus der folgenden Packung einsetzender Durchbruch- bzw. Schmierblutungen (wie beim Verschieben der Entzugsblutung).
4.3 Gegenanzeigen
Kombinierte hormonale Kontrazeptiva (KHK) dürfen unter den folgenden Bedingungen nicht angewendet werden:
• Überempfindlichkeit gegen die Wirkstoffe oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile
• Vorliegen einer oder Risiko für eine venöse Thromboembolie (VTE)
o Venöse Thromboembolie - bestehende VTE (auch unter Therapie mit Antikoagulanzien) oder VTE in der Vorgeschichte (z. B. tiefe Venenthrombose [TVT] oder Lungenembolie [LE]) o Bekannte erbliche oder erworbene Prädisposition für eine venöse Thromboembolie, wie z. B. APC-Resistenz (einschließlich Faktor-V-Leiden), Antithrombin-III-Mangel, Protein-C-Mangel oder Protein-S-Mangel
o Größere Operationen mit längerer Immobilisierung (siehe Abschnitt 4.4) o Hohes Risiko für eine venöse Thromboembolie aufgrund mehrerer Risikofaktoren (siehe Abschnitt 4.4)
• Vorliegen einer oder Risiko für eine arterielle Thromboembolie (ATE)
o Arterielle Thromboembolie - bestehende ATE, ATE in der Vorgeschichte (z. B.
Myokardinfarkt) oder Erkrankung im Prodromalstadium (z. B. Angina pectoris) o Zerebrovaskuläre Erkrankung - bestehender Schlaganfall, Schlaganfall oder prodromale Erkrankung (z. B. transitorische ischämische Attacke [TIA]) in der Vorgeschichte o Bekannte erbliche oder erworbene Prädisposition für eine arterielle Thromboembolie, wie z. B. Hyperhomocysteinämie und Antiphospholipid-Antikörper (Anticardiolipin-Antikörper, Lupusantikoagulans)
o Migräne mit fokalen neurologischen Symptomen in der Vorgeschichte o Hohes Risiko für eine arterielle Thromboembolie aufgrund mehrerer Risikofaktoren (siehe Abschnitt 4.4) oder eines schwerwiegenden Risikofaktors wie:
• Diabetes mellitus mit Gefäßschädigung
• Schwere Hypertonie
• Schwere Dyslipoproteinämie
• Bestehende oder vorausgegangene schwere Lebererkrankung, solange sich die Leberfunktionswerte nicht normalisiert haben
• Schwere Niereninsuffizienz oder akutes Nierenversagen
• Bestehende oder vorausgegangene Lebertumoren (benigne oder maligne)
• Bekannte oder vermutete sexualhormonabhängige, maligne Tumoren (z. B. der Genitalorgane oder der Brust)
• Diagnostisch nicht abgeklärte vaginale Blutungen.
4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung Warnhinweise
Die Eignung von Drosfemine sollte mit der Frau besprochen werden, falls eine der im Folgenden aufgeführten Erkrankungen oder Risikofaktoren vorliegt.
Bei einer Verschlechterung oder dem ersten Auftreten einer dieser Erkrankungen oder Risikofaktoren ist der Anwenderin anzuraten, sich an Ihren Arzt zu wenden, um zu entscheiden, ob die Anwendung von Drosfemine beendet werden sollte.
Schwerwiegende Nebenwirkungen kombinierter hormonaler Kontrazeptiva Risiko für eine venöse Thromboembolie (VTE)
Die Anwendung jedes kombinierten hormonalen Kontrazeptivums (KHK) erhöht das Risiko für eine venöse Thromboembolie (VTE) im Vergleich zur Nichtanwendung. Arzneimittel, die Levonorgestrel, Norgestimat oder Norethisteron enthalten, sind mit dem geringsten Risiko für eine VTE verbunden. Andere Arzneimittel, wie Drosfemine, können ein bis zu doppelt so hohes Risiko aufweisen. Die Entscheidung, ein Arzneimittel anzuwenden, das nicht zu denen mit dem geringsten VTE-Risiko gehört, sollte nur nach einem Gespräch mit der Frau getroffen werden, bei dem sicherzustellen ist, dass sie Folgendes versteht: das Risiko für eine VTE bei Anwendung von Drosfemine, wie ihre vorliegenden individuellen Risikofaktoren dieses Risiko beeinflussen und dass ihr Risiko für VTE in ihrem allerersten Anwendungsjahr am höchsten ist. Es gibt zudem Hinweise, dass das Risiko erhöht ist, wenn die Anwendung eines KHK nach einer Unterbrechung von 4 oder mehr Wochen wieder aufgenommen wird.
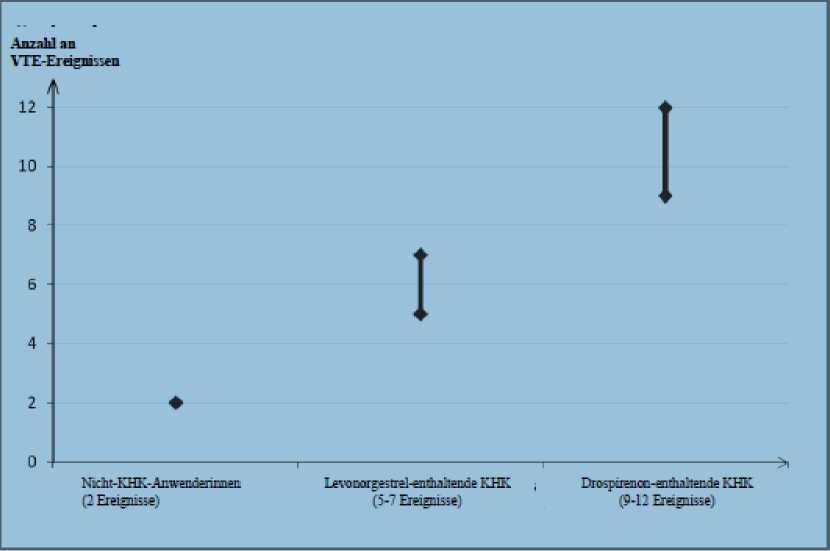
Ungefähr 2 von 10.000 Frauen, die kein KHK anwenden und nicht schwanger sind, erleiden im Verlauf eines Jahres eine VTE. Bei einer einzelnen Frau kann das Risiko jedoch in Abhängigkeit von ihren zugrunde liegenden Risikofaktoren bedeutend höher sein (siehe unten).
Es wird geschätzt1, dass im Verlauf eines Jahres 9 bis 12 von 10.000 Frauen, die ein Drospirenon-haltiges KHK anwenden, eine VTE erleiden; im Vergleich hierzu kommt es pro Jahr bei ungefähr 62 von 10.000 Frauen, die ein Levonorgestrel-haltiges KHK anwenden, zu einer VTE.
In beiden Fällen ist die Anzahl an VTE pro Jahr geringer als die erwartete Anzahl während der Schwangerschaft oder in der Zeit nach der Geburt.
VTE verlaufen in 1-2 % der Fälle tödlich.
Levonorgestrel-haltige KHK versus Nichtanwendung von ungefähr 2,3 bis 3,6.
Jährliche Anzahl an VTE-Ereignissen pro 10.000 Frauen
Äußerst selten wurde bei Anwenderinnen von KHK über eine Thrombose in anderen Blutgefäßen berichtet, wie z. B. in Venen und Arterien von Leber, Mesenterium, Nieren oder Retina.
Risikofaktoren für VTE
Das Risiko für venöse thromboembolische Komplikationen bei Anwenderinnen von KHK kann deutlich ansteigen, wenn bei der Anwenderin zusätzliche Risikofaktoren bestehen, insbesondere wenn mehrere Risikofaktoren vorliegen (siehe Tabelle).
Drosfemine ist kontraindiziert, wenn bei einer Frau mehrere Risikofaktoren gleichzeitig bestehen, die sie insgesamt einem hohen Risiko für eine Venenthrombose aussetzen (siehe Abschnitt 4.3). Weist eine Frau mehr als einen Risikofaktor auf, ist es möglich, dass der Anstieg des Risikos das Risiko der Summe der einzelnen Faktoren übersteigt - in diesem Fall muss ihr Gesamtrisiko für eine VTE in Betracht gezogen werden. Wenn das Nutzen/Risiko-Verhältnis als ungünstig erachtet wird, darf ein KHK nicht verschrieben werden (siehe Abschnitt 4.3).
Tabelle: Risikofaktoren für VTE
|
Risikofaktor |
Anmerkung |
|
Adipositas (Body-Mass-Index über 30 kg/m2) |
Das Risiko nimmt mit steigendem BMI deutlich zu. Besonders wichtig, wenn weitere Risikofaktoren vorliegen. |
|
Längere Immobilisierung, größere Operationen, jede Operation an Beinen oder Hüfte, neurochirurgische Operation oder schweres Trauma |
In diesen Fällen ist es ratsam, die Anwendung der Tablette (bei einer geplanten Operation mindestens vier Wochen vorher) zu unterbrechen und erst zwei Wochen nach der kompletten Mobilisierung wieder aufzunehmen. Es ist eine andere Verhütungsmethode anzuwenden, um eine ungewollte Schwangerschaft zu verhindern. |
|
Hinweis: Eine vorübergehende Immobilisierung einschließlich einer Flugreise von > 4 Stunden Dauer kann ebenfalls einen Risikofaktor für eine VTE darstellen, insbesondere bei Frauen mit weiteren Risikofaktoren. |
Eine antithrombotische Therapie muss erwogen werden, wenn Drosfemine nicht vorab abgesetzt wurde. |
|
Familiäre Vorbelastung (jede venöse Thromboembolie bei einem Geschwister- oder Elternteil, insbesondere in relativ jungen Jahren, z. |
Bei Verdacht auf eine genetische Prädisposition ist die Frau zur Beratung an einen Spezialisten zu überweisen, bevor eine Entscheidung über die Anwendung eines KHKs getroffen wird. |
|
Andere Erkrankungen, die mit einer VTE verknüpft sind |
Krebs, systemischer Lupus erythematodes, hämolytisches urämisches Syndrom, chronisch entzündliche Darmerkrankung (Morbus Crohn oder Colitis ulcerosa) und Sichelzellkrankheit |
|
Zunehmendes Alter |
Insbesondere älter als 35 Jahre |
Es besteht kein Konsens über die mögliche Rolle von Varizen und oberflächlicher Thrombophlebitis bezüglich des Beginns oder Fortschreitens einer Venenthrombose.
Das erhöhte Risiko einer Thromboembolie in der Schwangerschaft und insbesondere während der 6-wöchigen Dauer des Wochenbetts muss berücksichtigt werden (Informationen zu Schwangerschaft und Stillzeit siehe Abschnitt 4.6).
Symptome einer VTE (tiefe Beinvenenthrombose und Lungenembolie)
Beim Auftreten von Symptomen ist den Anwenderinnen anzuraten, unverzüglich ärztliche Hilfe in Anspruch zu nehmen und das medizinische Fachpersonal darüber zu informieren, dass sie ein KHK anwenden.
Bei einer tiefen Beinvenenthrombose (TVT) können folgende Symptome auftreten:
- unilaterale Schwellung des Beins und/oder Fußes oder entlang einer Beinvene
- Schmerz oder Druckschmerz im Bein, der möglicherweise nur beim Stehen oder Gehen bemerkt wird
- Erwärmung des betroffenen Beins; gerötete oder entfärbte Haut am Bein.
Bei einer Lungenembolie (LE) können folgende Symptome auftreten:
- plötzliches Auftreten unerklärlicher Kurzatmigkeit oder schnellen Atmens
- plötzlich auftretender Husten möglicherweise in Verbindung mit Hämoptyse
- stechender Brustschmerz
- starke Benommenheit oder Schwindelgefühl
- schneller oder unregelmäßiger Herzschlag.
Einige dieser Symptome (z. B. „Kurzatmigkeit“, „Husten“) sind unspezifisch und können als häufiger vorkommende und weniger schwerwiegende Ereignisse fehlinterpretiert werden (z. B. als Atemwegsinfektionen).
Andere Anzeichen für einen Gefäßverschluss können plötzlicher Schmerz sowie Schwellung und leicht bläuliche Verfärbung einer Extremität sein.
Tritt der Gefäßverschluss im Auge auf, können die Symptome von einem schmerzlosen verschwommenen Sehen bis zu einem Verlust des Sehvermögens reichen. In manchen Fällen tritt der Verlust des Sehvermögens sehr plötzlich auf.
Risiko für eine arterielle Thromboembolie (ATE)
Epidemiologische Studien haben die Anwendung von KHK mit einem erhöhten Risiko für arterielle Thromboembolie (Myokardinfarkt) oder apoplektischen Insult (z. B. transitorische ischämische Attacke, Schlaganfall) in Verbindung gebracht. Arterielle thromboembolische Ereignisse können tödlich verlaufen.
Risikofaktoren für ATE
Das Risiko für arterielle thromboembolische Komplikationen oder einen apoplektischen Insult bei Anwenderinnen von KHK erhöht sich bei Frauen, die Risikofaktoren aufweisen (siehe Tabelle). Drosfemine ist kontraindiziert bei Frauen, die einen schwerwiegenden oder mehrere Risikofaktoren für eine ATE haben, die sie einem hohen Risiko für eine Arterienthrombose aussetzen (siehe Abschnitt 4.3). Weist eine Frau mehr als einen Risikofaktor auf, ist es möglich, dass der Anstieg des Risikos das Risiko der Summe der einzelnen Faktoren übersteigt- in diesem Fall muss ihr Gesamtrisiko betrachtet werden. Bei Vorliegen eines ungünstigen Nutzen/Risiko-Verhältnisses darf ein KHK nicht verschrieben werden (siehe Abschnitt 4.3).
Tabelle: Risikofaktoren für ATE
|
Risikofaktor |
Anmerkung |
|
Zunehmendes Alter |
Insbesondere älter als 35 Jahre |
|
Rauchen |
Frauen ist anzuraten, nicht zu rauchen, wenn sie ein KHK anwenden möchten. Frauen über 35 Jahren, die weiterhin rauchen, ist dringend zu empfehlen, eine andere Verhütungsmethode anzuwenden. |
|
Hypertonie | |
|
Adipositas (Body-Mass-Index über 30 kg/m2) |
Das Risiko nimmt mit steigendem BMI deutlich zu. Besonders wichtig bei Frauen mit zusätzlichen Risikofaktoren. |
|
Familiäre Vorbelastung (jede arterielle Thromboembolie bei einem Geschwister- oder Elternteil, insbesondere in relativ jungen Jahren, d. h. jünger als 50 Jahre) |
Bei Verdacht auf eine genetische Prädisposition ist die Frau zur Beratung an einen Spezialisten zu überweisen, bevor eine Entscheidung über die Anwendung eines KHKs getroffen wird. |
|
Migräne |
Ein Anstieg der Häufigkeit oder des Schweregrads der Migräne während der Anwendung von KHK (die einem zerebrovaskulären Ereignis vorausgehen kann) kann ein Grund für ein sofortiues Absetzen sein. |
|
Andere Erkrankungen, die mit unerwünschten Gefäßereignissen verknüpft sind |
Diabetes mellitus, Hyperhomocysteinämie, Erkrankung der Herzklappen und Vorhofflimmern, Dyslipoproteinämie und systemischer Lupus erythematodes |
Symptome einer ATE
Beim Auftreten von Symptomen ist den Frauen anzuraten, unverzüglich ärztliche Hilfe in Anspruch zu nehmen und das medizinische Fachpersonal darüber zu informieren, dass sie ein KHK anwenden.
Bei einem apoplektischen Insult können folgende Symptome auftreten:
- plötzliche Taubheitsgefühle oder Schwäche in Gesicht, Arm oder Bein, besonders auf einer Körperseite
- plötzliche Gehschwierigkeiten, Schwindelgefühl, Gleichgewichtsverlust oder Koordinationsstörungen
- plötzliche Verwirrtheit, Sprech- oder Verständnisschwierigkeiten
- plötzliche Sehstörungen in einem oder beiden Augen
- plötzliche, schwere oder länger anhaltende Kopfschmerzen unbekannter Ursache
- Verlust des Bewusstseins oder Ohnmacht mit oder ohne Krampfanfall.
Vorübergehende Symptome deuten auf eine transitorische ischämische Attacke (TIA) hin.
Bei einem Myokardinfarkt (MI) können folgende Symptome auftreten:
- Schmerz, Unbehagen, Druck, Schweregefühl, Enge- oder Völlegefühl in Brust, Arm oder unterhalb des Sternums
- in den Rücken, Kiefer, Hals, Arm, Magen ausstrahlende Beschwerden
- Völlegefühl, Indigestion oder Erstickungsgefühl
- Schwitzen, Übelkeit, Erbrechen oder Schwindelgefühl
- extreme Schwäche, Angst oder Kurzatmigkeit
- schnelle oder unregelmäßige Herzschläge.
Tumorerkrankungen
In einigen epidemiologischen Untersuchungen wurde über ein erhöhtes Zervixkarzinom-Risiko bei Langzeitanwendung von KOK (> 5 Jahre) berichtet. Kontrovers diskutiert wird nach wie vor, in welchem Ausmaß dieses Ergebnis durch das Sexualverhalten und andere Faktoren, wie eine Infektion mit dem humanen Papillomavirus (HPV), beeinflusst wird.
Eine Metaanalyse von 54 epidemiologischen Studien hat ein leicht erhöhtes Brustkrebsrisiko (RR = 1,24) bei Frauen ergeben, die aktuell KOK anwenden. Das erhöhte Risiko geht innerhalb von 10 Jahren nach dem Absetzen von KOK allmählich wieder zurück. Da Brustkrebs bei Frauen unter 40 Jahren selten auftritt, ist die Anzahl zusätzlicher Brustkrebserkrankungen bei Anwenderinnen von KOK oder solchen, die früher KOK eingenommen haben, gering im Vergleich zum Gesamtrisiko an Brustkrebs zu erkranken. Ein Kausalzusammenhang wurde mit diesen Studien nicht bewiesen. Das beobachtete erhöhte Risiko kann an einer früheren Diagnose des Brustkrebses bei KOK-Anwenderinnen, den biologischen Wirkungen von KOK oder einer Kombination beider Faktoren liegen. Brustkrebs, der bei Frauen diagnostiziert wird, die irgendwann einmal ein KOK angewendet haben, scheint klinisch weniger weit fortgeschritten zu sein als Krebs bei Frauen, die noch nie ein KOK angewendet haben.
In seltenen Fällen sind bei Anwenderinnen von KOK gutartige und noch seltener bösartige Lebertumoren beobachtet worden. In Einzelfällen führten diese Tumoren zu lebensbedrohlichen intraabdominellen Blutungen. Wenn starke Oberbauchbeschwerden, eine Lebervergrößerung oder Anzeichen einer intraabdominellen Blutung bei Frauen auftreten, die KOK einnehmen, sollte ein Lebertumor in die differentialdiagnostischen Überlegungen einbezogen werden.
Mit der Anwendung höher dosierter KOK (50 pg Ethinylestradiol) verringert sich das Risiko eines Endometrium- und Ovarialkarzinoms. Ob dies auch für niedriger dosierte KOK gilt, muss noch nachgewiesen werden.
Sonstige Erkrankungen
Die Gestagenkomponente in Drosfemine ist ein Aldosteronantagonist mit kaliumsparenden Eigenschaften. In den meisten Fällen wird kein Anstieg des Kaliumspiegels erwartet. Jedoch wurde in einer klinischen Studie bei manchen Patientinnen mit leichter bis mittlerer Einschränkung der Nierenfunktion bei der gleichzeitigen Einnahme von Drospirenon und einem kaliumsparenden Arzneimittel ein leichter, aber nicht signifikanter Anstieg des Kaliumspiegels beobachtet. Aus diesem Grund ist es ratsam, bei Patientinnen mit Niereninsuffizienz und Serumkaliumwerten im oberen Referenzbereich vor der Behandlung und insbesondere bei gleichzeitiger Einnahme von kaliumsparenden Arzneimitteln die Serumkaliumwerte im ersten Behandlungszyklus zu kontrollieren. Siehe auch Abschnitt 4.5.
Frauen mit einer Hypertriglyzeridämie oder einer diesbezüglich positiven Familienanamnese können ein erhöhtes Risiko für die Entwicklung einer Pankreatitis haben, wenn sie KOK einnehmen.
Obwohl bei vielen Frauen, die KOK einnehmen, ein geringer Blutdruckanstieg berichtet wurde, sind klinisch relevante Blutdruckerhöhungen selten. Nur in diesen seltenen Fällen ist eine sofortige Unterbrechung der Einnahme des KOKs gerechtfertigt. Wenn es bei einer bereits existierenden Hypertonie und der gleichzeitigen Einnahme eines KOKs zu ständig erhöhten Blutdruckwerten oder einer signifikanten Erhöhung des Blutdrucks kommt, und in diesen Fällen eine antihypertensive Therapie keine Wirkung zeigt, muss das KOK abgesetzt werden. Wenn es angemessen erscheint, kann die Einnahme des KOKs wieder begonnen werden, sobald sich die Blutdruckwerte unter der antihypertensiven Therapie normalisiert haben.
Die folgenden Erkrankungen sollen Berichten zufolge sowohl in der Schwangerschaft als auch unter Anwendung eines KOKs auftreten bzw. sich verschlechtern, jedoch konnte ein Zusammenhang mit der Anwendung von KOK nicht bewiesen werden: cholestatischer Ikterus und/oder Pruritus, Gallensteine, Porphyrie, systemischer Lupus erythematodes, hämolytisch-urämisches Syndrom, Sydenham-Chorea, Herpes gestationis, Otosklerose-bedingte Schwerhörigkeit.
Bei Frauen mit hereditärem Angioödem können exogen zugeführte Estrogene Symptome eines Angioödems auslösen oder verschlimmern.
Akute oder chronische Leberfunktionsstörungen können eine Unterbrechung der Anwendung des KOKs erforderlich machen, bis sich die Leberfunktionswerte wieder normalisiert haben. Auch ein Rezidiv eines in einer vorausgegangenen Schwangerschaft oder während einer früheren Anwendung von steroidalen Geschlechtshormonen aufgetretenen cholestatischen Ikterus und/oder eines cholestasebedingten Pruritus macht das Absetzen des KOKs erforderlich.
Obwohl KOK einen Einfluss auf die periphere Insulinresistenz und Glucosetoleranz haben können, liegen keine Hinweise auf die Notwendigkeit einer Änderung des Therapieregimes bei Diabetikerinnen vor, die niedrig dosierte KOK anwenden (mit < 50 pg Ethinylestradiol). Diabetikerinnen müssen jedoch sorgfältig überwacht werden, insbesondere in der ersten Zeit der Anwendung eines KOKs.
Bei Anwendung von KOK wurde über eine Verschlechterung von endogenen Depressionen, Epilepsie, Morbus Crohn und Colitis ulcerosa berichtet.
Chloasmen können gelegentlich auftreten, insbesondere bei Frauen mit Chloasma gravidarum in der Anamnese. Frauen mit dieser Veranlagung sollten sich daher während der Einnahme von KOK nicht direkt der Sonne oder ultraviolettem Licht aussetzen.
Drosfemine enthält Lactose. Anwenderinnen mit der seltenen hereditären Galactose-Intoleranz, Lapp-Lactase-Mangel oder Glucose-Galactose-Malabsorption sollten dieses Arzneimittel nicht einnehmen.
Ärztliche Untersuchung/Beratung
Vor der Einleitung oder Wiederaufnahme der Behandlung mit Drosfemine muss eine vollständige Anamnese (inklusive Familienanamnese) erhoben und eine Schwangerschaft ausgeschlossen werden. Der Blutdruck sollte gemessen und eine körperliche Untersuchung durchgeführt werden, die sich an den Gegenanzeigen (siehe Abschnitt 4.3) und Warnhinweisen (siehe Abschnitt 4.4) orientiert. Es ist wichtig, die Frau auf die Informationen zu venösen und arteriellen Thrombosen hinzuweisen, einschließlich des Risikos von Drosfemine im Vergleich zu anderen KHK, die Symptome einer VTE und ATE, die bekannten Risikofaktoren und darauf, was im Falle einer vermuteten Thrombose zu tun ist.
Die Anwenderin ist zudem anzuweisen, die Packungsbeilage sorgfältig zu lesen und die darin gegebenen Ratschläge zu befolgen. Die Häufigkeit und Art der Untersuchungen sollte den gängigen Untersuchungsleitlinien entsprechen und individuell auf die Frau abgestimmt werden.
Die Anwenderinnen sind darüber aufzuklären, dass hormonale Kontrazeptiva nicht vor HIV-Infektionen (AIDS) und anderen sexuell übertragbaren Krankheiten schützen.
Verminderte Wirksamkeit
Die Wirksamkeit von KOK kann beeinträchtigt sein, wenn z. B. wirkstoffhaltige Tabletten vergessen wurden (siehe Abschnitt 4.2), bei gastrointestinalen Beschwerden während der Einnahme wirkstoffhaltiger Tabletten (siehe Abschnitt 4.2) oder wenn gleichzeitig bestimmte andere Arzneimittel eingenommen werden (siehe Abschnitt 4.5).
Unregelmäßige Blutungen
Bei allen KOK kann es, insbesondere in den ersten Monaten der Anwendung, zu unregelmäßigen Blutungen (Schmier- oder Durchbruchblutungen) kommen. Eine diagnostische Abklärung dieser Zwischenblutungen ist deshalb erst nach einer Anpassungsphase von ungefähr drei Zyklen sinnvoll.
Bei anhaltenden, unregelmäßigen Blutungen oder beim Auftreten von Blutungsunregelmäßigkeiten bei bislang regelmäßigen Zyklen sollten nichthormonale Ursachen in Betracht gezogen und entsprechende diagnostische Maßnahmen ergriffen werden, um eine maligne Erkrankung oder eine Schwangerschaft auszuschließen. Dies kann auch eine Kürettage beinhalten.
Es ist möglich, dass es bei einigen Anwenderinnen in der Placebotablettenphase zu keiner Entzugsblutung kommt.
Wenn das KOK wie unter Abschnitt 4.2 beschrieben eingenommen wurde, ist eine Schwangerschaft unwahrscheinlich. Wenn die Einnahme des KOKs jedoch vor der ersten ausgebliebenen Entzugsblutung nicht vorschriftsmäßig erfolgt ist oder bereits zum zweiten Mal die Entzugsblutung ausgeblieben ist, muss eine Schwangerschaft ausgeschlossen werden, bevor die Anwendung des KOKs fortgesetzt wird.
4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Hinweis: Die Fachinformationen gleichzeitig verordneter Arzneimittel sollten auf mögliche Wechselwirkungen überprüft werden.
Wirkung anderer Arzneimittel auf Drosfemine
Es können Wechselwirkungen mit Arzneimitteln vorkommen,die mikrosomale Enzyme induzieren. Dies kann eine erhöhte Clearance von Sexualhormonen zur Folge haben und zu Durchbruchblutungen und/oder Versagen des oralen Kontrazeptivums führen.
• Vorgehensweise
Eine Enzyminduzierung kann bereits nach wenigen Tagen der Behandlung beobachtet werden. Der maximale enzyminduzierende Effekt wird üblicherweise innerhalb weniger Wochen beobachtet. Nach Beendigung der Therapie kann der enzyminduzierende Effekt noch bis zu 4 Wochen anhalten.
• Kurzzeitige Behandlung
Frauen, die mit Leberenzym-induzierenden Arzneimitteln behandelt werden, sollten vorübergehend eine Barrieremethode oder eine andere Verhütungsmethode zusätzlich zu dem KOK anwenden. Die Barrieremethode muss während der gesamten Dauer der gleichzeitigen Anwendung der Arzneimittel und bis zu 28 Tage nach Absetzen der Behandlung verwendet werden. Wenn eines dieser Arzneimittel auch dann noch weiter eingenommen werden muss, nachdem die aktiven Tabletten der KOK-Packung verbraucht sind, müssen die Placebo-Tabletten verworfen werden und mit der Einnahme aus der nächsten Blisterpackung des KOK begonnen werden.
• Langzeitbehandlung
Bei Frauen, die langfristig mit Leberenzym-induzierenden Wirkstoffen behandelt werden, wird die Anwendung einer anderen zuverlässigen, nichthormonalen Verhütungsmethode empfohlen.
Die folgenden Wechselwirkungen sind in der Literatur beschrieben.
Substanzen, die die Clearance von KOK erhöhen (verminderte Wirksamkeit von KOK durch Enzyminduktion) wie z.B.:
Barbiturate, Bosentan, Carbamazepin, Phenytoin, Primidon, Rifampicin und die zur Behandlung einer HIV-Infektion verwendeten Arzneimittel Ritonavir, Nevirapin und Efavirenz und möglicherweise auch Felbamat, Griseofulvin, Oxcarbazepin, Topiramat und Produkte, die das pflanzliche Heilmittel Johanniskraut (Hypericum perforatum) enthalten.
Substanzen mit unterschiedlicher Wirkung auf die Clearance von KOK:
Viele HIV/HCV-Proteaseinhibitoren und nichtnukleosidale Reverse-Transkriptase-Hemmer können bei gleichzeitiger Verabreichung mit KOK die Plasmakonzentrationen von Estrogenen und Gestagenen erhöhen oder senken. Diese Veränderungen können in einigen Fällen klinisch relevant sein. Daher sollten die Fachinformationen der gleichzeitig verordneten HIV/HCV-Arzneimittel auf mögliche Wechselwirkungen und damit verbundene Empfehlungen überprüft werden. Im Zweifel sollte von Frauen während der Therapie mit Proteaseinhibitoren oder nichtnukleosidale Reverse-Transkriptase-Hemmern eine zusätzliche Barrieremethode zur Empfängnisverhütung verwendet werden.
Die Hauptmetaboliten von Drospirenon im menschlichen Plasma entstehen ohne Beteiligung des Cytochrom-P450-Systems. Es ist daher unwahrscheinlich, dass Inhibitoren dieses Enzymsystems den Metabolismus von Drospirenon beeinflussen.
Wirkung von Drosfemine auf andere Arzneimittel
Orale Kontrazeptiva können den Stoffwechsel bestimmter anderer Wirkstoffe beeinflussen. Entsprechend können Plasma- und Gewebekonzentrationen entweder erhöht (z. B. Ciclosporin) oder erniedrigt (z. B. Lamotrigin) werden.
In-vitro-Hemmstudien und In-vivo-Studien zu Wechselwirkungen bei Frauen, die Omeprazol, Simvastatin und Midazolam als Markersubstrat erhielten, zeigten, dass eine Wechselwirkung von Drospirenon in Dosen von 3 mg mit dem Metabolismus anderer Wirkstoffe unwahrscheinlich ist.
Sonstige Arten von Wechselwirkungen
Bei nierengesunden Patientinnen zeigte die gleichzeitige Einnahme von Drospirenon und ACE-Hemmern oder NSARs keinen signifikanten Effekt auf den Kaliumspiegel im Serum. Die gleichzeitige Einnahme von Drosfemine und Aldosteronantagonisten oder kaliumsparenden Diuretika wurde jedoch nicht untersucht. In diesem Fall sollte im ersten Behandlungszyklus das Serumkalium kontrolliert werden (siehe auch Abschnitt 4.4).
Laboruntersuchungen
Die Anwendung von steroidalen Kontrazeptiva kann die Ergebnisse bestimmter Labortests beeinflussen, u. a. die biochemischen Parameter der Leber-, Schilddrüsen-, Nebennieren- und Nierenfunktion sowie die Plasmaspiegel von (Träger-)Proteinen, z. B. des kortikosteroidbindenden Globulins und der Lipid-/Lipoprotein-Fraktionen, die Parameter des Kohlenhydratstoffwechsels sowie die Gerinnungs- und Fibrinolyseparameter. Im Allgemeinen bleiben diese Veränderungen jedoch innerhalb des Normbereichs. Drospirenon führt aufgrund seiner leichten antimineralokortikoiden Wirkung zu einem Anstieg der Reninaktivität im Plasma und des Plasmaaldosterons.
4.6 Fertilität, Schwangerschaft und Stillzeit
Schwangerschaft
Drosfemine ist während einer Schwangerschaft nicht indiziert.
Falls unter der Einnahme von Drosfemine eine Schwangerschaft eintritt, ist das Arzneimittel sofort abzusetzen. In umfangreichen epidemiologischen Untersuchungen fand sich weder ein erhöhtes Risiko für Missbildungen bei Kindern, deren Mütter vor der Schwangerschaft KOK eingenommen hatten, noch eine teratogene Wirkung bei versehentlicher Einnahme von KOK in der Schwangerschaft.
Tierstudien zeigten unerwünschte Wirkungen während der Trächtigkeit und Laktation (siehe Abschnitt 5.3). Aufgrund dieser Ergebnisse bei Tieren können hormonelle Nebenwirkungen der Wirkstoffe nicht ausgeschlossen werden. Allgemeine Erfahrungen mit KOK während der Schwangerschaft ergaben jedoch keine Hinweise auf unerwünschte Wirkungen beim Menschen.
Die verfügbaren Daten zur Anwendung von Drosfemine während der Schwangerschaft sind zu begrenzt, um Schlussfolgerungen hinsichtlich negativer Auswirkungen von Drosfemine auf die Schwangerschaft und die Gesundheit des Fetus oder des Neugeborenen zu ermöglichen. Bisher sind keine einschlägigen epidemiologischen Daten verfügbar.
Das erhöhte VTE-Risiko in der Zeit nach der Geburt sollte vor der erneuten Anwendung nach einer Anwendungspause bedacht werden (siehe Abschnitte 4.2 und 4.4).
Stillzeit
KOK können die Laktation beeinflussen, da sie die Menge der Muttermilch vermindern und ihre Zusammensetzung verändern können. Daher wird die Anwendung von KOK generell nicht empfohlen, solange eine Mutter ihr Kind nicht vollständig abgestillt hat. Geringe Mengen der kontrazeptiven Steroide und/oder ihrer Metaboliten können während der KOK-Anwendung in die Muttermilch ausgeschieden werden. Diese Mengen könnten das Kind beeinträchtigen.
Fertilität
Drosfemine ist indiziert für die Verhinderung der Schwangerschaft. Zu Informationen über die Rückkehr der Fruchtbarkeit siehe Abschnitt 5.1
4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Es wurden keine Studien zu den Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen durchgeführt. Es wurden keine Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen bei Anwenderinnen von KOK beobachtet.
4.8 Nebenwirkungen
Zu schweren Nebenwirkungen bei Anwenderinnen von KOK siehe Abschnitt 4.4.
Bei der Anwendung von 0,02 mg/3 mg Ethinylestradiol/Drospirenon, Dosisschema 24+4 Tage wurde über folgende Nebenwirkungen des Arzneimittels berichtet:
In der nachfolgenden Tabelle wurden die Nebenwirkungen nach MedDRA-Systemorganklassen kategorisiert (MedDRA SOCs). Die Häufigkeiten basieren auf Daten klinischer Studien. Der geeignetste MedDRA-Begriff wurde verwendet, um eine bestimmte Reaktion, ihre Synonyme und in Zusammenhang stehende Erkrankungen zu beschreiben.
Nebenwirkungen in Verbindung mit der Anwendung von 0,02 mg/3 mg Ethinylestradiol/Drospirenon, Dosisschema 24+4 Tage als orales Kontrazeptivum oder zur Behandlung von mäßiger Akne vulgaris nach den MedDRA-Systemorganklassen und den MedDRA-Begriffen
|
Organsystemklasse (MeDRA version 9.1) |
Häufigkeit von unerwünschten Wirkungen | |||
|
Häufig (> 1/100, <1/10) |
Gelegentlich (>1/1.000,<1/100) |
Selten (>1/10.000, <1/1.000) |
Nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar)> | |
|
Infektionen und parasitäre Erkrankungen |
Candidose | |||
|
Erkrankungen des Blutes und des Lymphsystems |
Anämie, Thrombozythämie | |||
|
Erkrankungen des Immunsystems |
Allergische Reaktionen |
Überempfindlichkeitsrea ktionen | ||
|
Endokrine Erkrankungen |
Endokrine Erkrankungen | |||
|
Stoffwechsel- und Ernährungsstörungen |
Zunahme des Appetits, Anorexie, Hyperkaliämie, Hyponatriämie | |||
|
Psychiatrische Erkrankungen |
Stimmungsschw ankungen |
Depression, Nervosität, Somnolenz |
Anorgasmie, Insomnie | |
|
Erkrankungen des Nervensystems |
Kopfschmerzen |
Benommenheit, Parästhesie |
Schwindel, Tremor | |
|
Augenerkrankungen |
Konjunktivitis, Augentrockenheit, Augenerkrankungen | |||
|
Herzerkrankungen |
Tachykardie | |||
|
Gefäßerkrankungen |
Migräne, Varizen, |
Phlebitis, | ||
|
Hypertonie |
Gefäßerkrankung, Epistaxis, Synkope, VTE/ATE | |||
|
Erkrankungen des Gastrointestinaltrakts |
Übelkeit |
Bauchschmerzen, Erbrechen, Dyspepsie, Flatulenz, Gastritis, Diarrhö |
Abdomenvergrößerun g, Gastrointestinale Erkrankung, Gastrointestinales Völlegefühl, Hiatushernie, Orale Candidose, Obstipation, Mundtrockenheit | |
|
Leber- und Gallenerkrankungen |
Gallenschmerzen, Cholezystitis | |||
|
Erkankungen der Haut und des U nterhautzellgewebe s |
Akne, Pruritus, Hautauschlag |
Chloasma, Ekzem, Alopezie, Akneforme Dermatitis, Trockene Haut, Erythema nodosum, Hypertrichiosis, Erkrankungen der Haut, Striae der Haut, Kontaktdermatitis, Photosensible Dermatitis, Hautknötchen |
Erythema multiforme | |
|
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankunge n |
Rückenschmerzen , Schmerzen in den Extremitäten, Muskelkrämpfe | |||
|
Erkrankungen der Geschlechtsorgane und der Brustdrüse |
Brustschmerzen, Metrorrhagie*, Amenorrhö |
Vaginale Candidose, Beckenschmerzen , Vergrößerung der Brust, Fibrozystische Brust, Uterus- /Vaginalblutunge n*, Genitalfluor, Hitzewallungen, Vaginitis, Zyklusstörungen, Dysmenorrhö, Hypomenorrhö, Menorrhagie, Vaginale Trockenheit, Auffälliger Papanicolaou- Abstrich, Abnahme der Libido |
Dyspareunie, Vulvovaginitis, Postkoitale Blutung, Entzugsblutung, Brustzysten, Brusthyperplasie, Neoplasie der Brust, Zervixpolyp, Endometriumatrophie , Ovarialzysten, Vergrößerung des Uterus | |
|
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort |
Asthenie, Vermehrtes Schwitzen, Ödeme (Ödemkrankheit, peripheres Ödem, |
Unwohlsein |
|
Gesichtsödem) | ||||
|
Untersuchungen |
Gewichtszunahme |
Gewichtsabnahme |
* Unregelmäßige Blutungen lassen während fortgesetzter Behandlung üblicherweise nach
Bei Anwenderinnen von KOK wurde über die folgenden schweren unerwünschten Ereignisse berichtet, die in Abschnitt 4.4 „Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung“ erläutert werden:
- Hypertonie
- Lebertumoren
- Auftreten oder Verschlechterung von Erkrankungen, für die ein Zusammenhang mit der Einnahme von KOK nicht eindeutig nachgewiesen ist: Morbus Crohn, Colitis ulcerosa, Epilepsie, Uterusmyome, Porphyrie, systemischer Lupus erythematodes, Herpes gestationis, Sydenham-Chorea, hämolytisch urämisches Syndrom, cholestatischer Ikterus
- Chloasma
- Akute oder chronische Leberfunktionsstörungen können die Unterbrechung der Einnahme von KOK erforderlich machen, bis sich die Leberfunktionswerte wieder normalisiert haben.
- Bei Frauen mit hereditärem Angioödem können exogen zugeführte Estrogene Symptome eines Angioödems auslösen oder verschlimmern.
Beschreibung ausgewählter Nebenwirkungen
Bei Anwenderinnen von KHK wurde ein erhöhtes Risiko für arterielle und venöse thrombotische und thromboembolische Ereignisse einschließlich Myokardinfarkt, Schlaganfall, transitorische ischämische Attacken, Venenthrombose und Lungenembolie beobachtet, die in Abschnitt 4.4 eingehender behandelt werden.
Die Diagnosehäufigkeit von Brustkrebs unter Anwenderinnen von KOK ist geringfügig erhöht. Da bei Frauen unter 40 Jahren Brustkrebs selten auftritt, ist das zusätzliche Risiko, an Brustkrebs zu erkranken, im Verhältnis zum Gesamtrisiko gering. Die Kausalität mit der Anwendung von KOK ist nicht bekannt. Für weitere Informationen siehe Abschnitte 4.3 und 4.4.
Wechselwirkungen
Durchbruchblutungen und/oder Versagen der kontrazeptiven Wirkung können aufgrund von Wechselwirkungen von KOK mit anderen Arzneimitteln (enzyminduzierende Arzneimittel) auftreten (siehe Abschnitt 4.5.).
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Arzneimittel und Medizinprodukte, Abt. Pharmakovigilanz, Kurt-Georg-Kiesinger-Allee 3, D-53175 Bonn, Website: www.bfarm.de anzuzeigen.
4.9 Überdosierung
Für Drosfemine liegen bislang keine Erfahrungen zur Überdosierung vor.
Ausgehend von den mit kombinierten oralen Kontrazeptiva gesammelten allgemeinen Erfahrungen können möglicherweise folgende Symptome auftreten, wenn eine Überdosis an wirkstoffhaltigen Tabletten eingenommen wurde: Übelkeit, Erbrechen und bei jungen Mädchen leichte vaginale Blutungen.
Es gibt kein Antidot und die weitere Behandlung erfolgt symptomatisch.
5. PHARMAKOLOGISCHE EIGENSCHAFTEN
Pharmakotherapeutische Gruppe: Gestagene und Estrogene, fixe Kombinationen ATC-Code: G03AA12
Pearl-Index für Methodenversagen: 0,41 (oberes zweiseitiges 95 %-Konfidenzlimit: 0,85) Gesamt-Pearl-Index (Methodenversagen + Anwendungsfehler): 0,80 (oberes zweiseitiges 95 %-Konfidenzlimit: 1,30)
Die kontrazeptive Wirkung von Drosfemine beruht auf dem Zusammenspiel verschiedener Faktoren, wobei die Ovulationshemmung und Endometriumveränderungen als die wichtigsten Faktoren anzusehen sind.
In einer Ovulationshemmstudie über 3 Zyklen, die 3 mg Drospirenon/ 0,02 mg Ethinylestradiol in einem 24-Tage-Regime und einem 21-Tage-Regime verglich, war das 24-Tage-Regime mit einer stärkeren Unterdrückung der follikulären Entwicklung verbunden. Nach absichtlichen Einnahmefehlern im dritten Zyklus der Einnahme zeigten anteilmäßig mehr Frauen unter dem 21-Tage-Regime eine ovarielle Aktivität einschließlich Ovulationen („escape ovulations“) als Frauen unter dem 24-Tage-Regime.
Die ovarielle Aktivität kehrte bei 91,8 % der Frauen, die das 24-Tage-Regime anwendeten, im Zyklus nach Einnahme („post-treatment“) wieder zu den Werten vor der Einnahme („pre-treatment“) zurück.
Drosfemine ist ein kombiniertes orales Kontrazeptivum mit Ethinylestradiol und dem Gestagen Drospirenon. In therapeutischer Dosierung hat Drospirenon auch antiandrogene und milde antimineralokortikoide Eigenschaften. Es zeigt keinerlei estrogene, glukokortikoide und antiglukokortikoide Wirkungen. Dies verleiht Drospirenon ein pharmakologisches Profil, das dem des natürlichen Hormons Progesteron sehr ähnlich ist.
Aus klinischen Studien geht hervor, dass die leichten antimineralokortikoiden Eigenschaften von Drosfemine zu einem leichten antimineralokortikoiden Effekt führen.
Zur Untersuchung der Wirksamkeit und Sicherheit von Drospirenon und Ethinylestradiol wurden zwei multizentrische, randomisierte, placebokontrollierte Doppelblindstudien an Frauen mit mäßiger Akne vulgaris durchgeführt.
Nach sechs monatiger Behandlung zeigte die kombinierte Anwendung von Drospirenon und Ethinylestradiol im Vergleich zu Placebo eine statistisch signifikant stärkere Verminderung von entzündlichen Läsionen um 15,6 % (49,3 % versus 33,7 %), von nicht-entzündlichen Läsionen um 18,5 % (40,6 % versus 22,1 %) sowie der Anzahl der Läsionen insgesamt um 16,5 % (44,6 % versus 28,1 %). Darüber hinaus erreichte ein um 11,8 % höherer prozentualer Anteil von Probandinnen (18,6 % versus 6,8 %) die Einstufung „rein“ oder „nahezu rein“ auf der Skala Investigator’s Static Global Assessment (ISGA).
5.2 Pharmakokinetische Eigenschaften
Drospirenon
Resorption
Oral angewendetes Drospirenon wird rasch und fast vollständig resorbiert. Nach einmaliger Einnahme werden nach ca. 1-2 Stunden maximale Wirkstoffkonzentrationen im Serum von etwa 38 ng/ml erreicht. Die Bioverfügbarkeit liegt zwischen 76 % und 85 %. Eine gleichzeitige Nahrungsaufnahme hat keinen Einfluss auf die Bioverfügbarkeit von Drospirenon.
Verteilung
Nach oraler Gabe sinken die Drospirenonspiegel im Serum mit einer terminalen Halbwertszeit von 31 Stunden. Drospirenon wird an Serumalbumin gebunden und bindet nicht an sexualhormonbindendes Globulin (SHBG) oder kortikosteroidbindendes Globulin (CBG). Nur 3 - 5 % der Gesamtkonzentration des Wirkstoffs im Serum liegen als freies Steroid vor. Der Ethinylestradiol-induzierte Anstieg des SHBG beeinflusst die Serumproteinbindung von Drospirenon nicht. Das mittlere scheinbare Verteilungsvolumen von Drospirenon beträgt 3,7 ± 1,2 l/kg.
Biotransformation
Drospirenon wird nach oraler Gabe weitgehend metabolisiert. Die Hauptmetaboliten im Plasma sind die Säureform von Drospirenon, die durch eine Öffnung des Lactonrings entsteht, und das 4,5-Dihydro-Drospirenon-3-sulfat. Beide Metaboliten werden ohne Beteiligung des P450-Systems gebildet. Drospirenon wird in geringem Ausmaß durch Cytochrom-P450 3A4 metabolisiert. In-vitro konnte gezeigt werden, dass Drospirenon Cytochrom-P450 3A4, Cytochrom-P450 1A1, Cytochrom-P450 2C9 und Cytochrom-P450 2C19 hemmt.
Elimination
Die metabolische Clearance-Rate von Drospirenon im Serum beträgt 1,5 ± 0,2 ml/min/kg. Drospirenon wird unverändert nur in Spuren ausgeschieden. Die Metaboliten von Drospirenon werden mit dem Stuhl und Urin bei einem Exkretionsverhältnis von ungefähr 1,2 bis 1,4 ausgeschieden. Die Halbwertszeit der Metabolitenausscheidung über Urin und Stuhl beträgt ungefähr 40 Stunden.
Steady-State-Bedingungen
Im Verlauf eines Behandlungszyklus werden die maximalen Steady-State-Konzentrationen von Drospirenon im Serum von ungefähr 70 ng/ml nach etwa 8 Behandlungstagen erreicht. Die Serumdrospirenonspiegel akkumulierten um einen Faktor von ungefähr 3 als Folge des Verhältnisses von terminaler Halbwertszeit und Dosisintervall.
Bestimmte Gruppen von Anwenderinnen
Auswirkungen einer Niereninsuffizienz
Die Steady-state-Serumspiegel von Drospirenon bei Frauen mit leichter Niereninsuffizienz (Kreatinin-Clearance CLcr, 50 - 80 ml/min) waren vergleichbar mit denen von Frauen mit normaler Nierenfunktion. Bei Frauen mit mäßiger Niereninsuffizienz (CLcr, 30 - 50 ml/min) waren die Serumspiegel von Drospirenon im Mittel um 37 % höher als bei Frauen mit normaler Nierenfunktion. Die Behandlung mit Drospirenon wurde auch von Frauen mit leichter bis mäßiger Niereninsuffizienz gut vertragen. Die Drospirenon-Behandlung hatte keinen klinisch signifikanten Einfluss auf die Kaliumkonzentration im Serum.
Auswirkungen einer Leberinsuffizienz
In einer Einzeldosisstudie nahm bei Probandinnen mit mäßiger Leberinsuffizienz die orale Clearance (CL/f) um ca. 50 % ab im Vergleich zu denen mit normaler Leberfunktion. Die beobachtete Abnahme der Drospirenon-Clearance bei Probandinnen mit mäßiger Leberinsuffizienz führte nicht zu erkennbaren Änderungen der Kaliumkonzentrationen im Serum. Selbst bei Diabetes und begleitender Behandlung mit Spironolacton (zwei prädisponierenden Faktoren für eine Hyperkaliämie) war kein Anstieg der Kaliumkonzentrationen im Serum über die obere Grenze des Normalbereichs zu beobachten. Es kann daher festgestellt werden, dass Drospirenon von Patienten mit leichter bis mäßiger Leberinsuffizienz (Child-Pugh B) gut vertragen wird.
Ethnische Gruppen
Zwischen japanischen und kaukasischen Frauen wurden keine klinisch bedeutsamen Unterschiede in der Pharmakokinetik von Drospirenon und Ethinylestradiol beobachtet.
Ethinylestradiol
Resorption
Oral angewendetes Ethinylestradiol wird schnell und vollständig resorbiert. Das Konzentrationsmaximum im Serum von ca. 33 pg/ml wird innerhalb von 1 - 2 Stunden nach oraler Einzelgabe erreicht. Infolge der präsystemischen Konjugation und des First-Pass-Metabolismus liegt die absolute Bioverfügbarkeit bei ca. 60 %. Bei ca. 25 % der untersuchten Probandinnen verringerte die gleichzeitige Nahrungsaufnahme die Bioverfügbarkeit von Ethinylestradiol, während bei den übrigen Probandinnen keine Änderung zu beobachten war.
Verteilung
Serumspiegel von Ethinylestradiol sinken in zwei Phasen. Die terminale Dispositionsphase ist von einer Halbwertszeit von ca. 24 Stunden gekennzeichnet. Ethinylestradiol wird stark (zu ca. 98,5 %), aber unspezifisch, an Serumalbumin gebunden. Ethinylestradiol induziert einen Anstieg der Konzentrationen von sexualhormonbindendem Globulin (SHBG) und kortikosteroidbindendem Globulin (CBG) im Serum. Das scheinbare Verteilungsvolumen wurde mit ca. 5 l/kg bestimmt.
Biotransformation
Ethinylestradiol unterliegt der präsystemischen Konjugation sowohl in der Mukosa des Dünndarms als auch in der Leber. Der primäre Metabolisierungsweg von Ethinylestradiol besteht in einer aromatischen Hydroxylierung. Jedoch werden zahlreiche hydroxylierte und methylierte Metabolite gebildet, die in freier Form und in Form von Glukuronid- und Sulfatkonjugaten vorliegen. Die metabolische Clearance-Rate von Ethinylestradiol beträgt ca. 5 ml/min/kg.
Elimination
Ethinylestradiol wird in keinem signifikanten Umfang in unveränderter Form ausgeschieden. Die Metaboliten von Ethinylestradiol werden über den Urin und die Galle im Verhältnis 4:6 ausgeschieden. Die Halbwertszeit der Metabolitenausscheidung liegt bei ungefähr 1 Tag.
Steady-State-Bedingungen
Steady-State-Bedingungen werden in der zweiten Hälfte des Behandlungszyklus erreicht und die Serumspiegel von Ethinylestradiol akkumulieren um einen Faktor von ungefähr 2,0 bis 2,3.
5.3 Präklinische Daten zur Sicherheit
In Tierversuchen beschränkten sich die Effekte von Drospirenon und Ethinylestradiol auf diejenigen, die mit den bekannten pharmakologischen Wirkungen in Verbindung gebracht werden. Insbesondere zeigten reproduktionstoxikologische Studien embryotoxische und fetotoxische Wirkungen bei den Versuchstieren, die als speziesspezifische Effekte bewertet wurden. Unter Exposition in Dosen, die über denen liegen, wie sie mit Drosfemine eingenommen werden, wurden Veränderungen in der Differenzierung der Sexualorgane von Rattenfeten, nicht aber bei Feten von Affen, beobachtet.
6. PHARMAZEUTISCHE ANGABEN 6.1 Liste der sonstigen Bestandteile
Wirkstoffhaltige Filmtabletten (rosa): Placebo-Filmtabletten(weiß):
Tablettenkern:
Lactose-Monohydrat Lactose-Monohydrat
Maisstärke Maisstärke
Magnesiumstearat (Ph.Eur.) [pflanzlich] Magnesiumstearat (Ph.Eur.) [pflanzlich]
Filmüberzug:
Hypromellose
Talkum
Titandioxid (E171) Polysorbat 80 Eisen(III)-oxid (E172)
Hypromellose Lactose-Monohydrat Titandioxid (E171) Macrogol 4000 Natriumcitrat (Ph.Eur.)
6.2 Inkompatibilitäten
Nicht zutreffend.
6.3 Dauer der Haltbarkeit
2 Jahre
6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Für dieses Arzneimittel sind keine besonderen Lagerungsbedingungen erforderlich.
6.5 Art und Inhalt des Behältnisses
Transparente PVC/PE/PVdC-Aluminium-Blisterpackung.
Packungsgrößen:
24+4 Tabletten
3 x 24+4 Tabletten 6 x 24+4 Tabletten 13 x 24+4 Tabletten
Jede Blisterpackung enthält 24 rosafarbene wirkstoffhaltige Filmtabletten und 4 weiße PlaceboFilmtabletten.
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung
Keine besonderen Anforderungen für die Beseitigung.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
7. INHABER DER ZULASSUNG
mibe GmbH Arzneimittel Münchener Straße 15 06796 Brehna Tel.: 034954/247-0 Fax: 034954/247-100
8. ZULASSUNGSNUMMER 89743.00.00
9. DATUM DER ERTEILUNG DER ZULASSUNG
27. August .2014
10. STAND DER INFORMATION
04.2015
11. VERKAUFSABGRENZUNG
V erschreibungspflichtig.
21
Diese Inzidenzen wurden aus der Gesamtheit der epidemiologischen Studiendaten abgeleitet, wobei relative Risiken der verschiedenen Arzneimittel im Vergleich zu Levonorgestrel-haltigen KHK verwendet wurden.
Mittelwert der Spannweite 5-7 pro 10.000 Frauenjahre, auf der Grundlage eines relativen Risikos für