Dysport 500 Einheiten, Pulver Zur Herstellung Einer Injektionslösung
Fachinformation (Zusammenfassung der Merkmale des Arzneimittels)
1. BEZEICHNUNG DES ARZNEIMITTELS
Dysport® 300 Einheiten / 500 Einheiten Pulver zur Herstellung einer Injektionslösung
2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
Eine Durchstechflasche enthält 300 bzw. 500 Einheiten ) Clostridium botulinum Toxin Typ A.
>j«\
) 1 Einheit entspricht der LD50 bei Mäusen nach intraperitonealer Injektion.
Die vollständige Auflistung der sonstigen Bestandteile siehe Abschnitt 6.1.
3. DARREICHUNGSFORM
Pulver zur Herstellung einer Injektionslösung
4. KLINISCHE ANGABEN
4.1 Anwendungsgebiete
• Zur symptomatischen Alternativbehandlung von idiopathischem Blepharospasmus, hemifazialem Spasmus und koexistierenden fokalen Dystonien.
• Zur symptomatischen Behandlung einer zervikalen Dystonie (Torticollis spasmodicus) mit Beginn im Erwachsenenalter.
• Zur symptomatischen Behandlung einer Armspastik bei Erwachsenen infolge eines Schlaganfalls. Hinweis
Der Patient ist vor Beginn einer Therapie mit Dysport darauf hinzuweisen, dass daneben noch andere (medikamentöse, chirurgische) Behandlungsmethoden bestehen, und dass nicht alle Patienten auf die Behandlung mit Dysport ansprechen bzw. nur eine partielle Symptomlinderung eintritt. Voraussagbare Faktoren für die nicht gegebene bzw. verminderte Ansprechbarkeit sind nicht bekannt.
4.2 Dosierung, Art und Dauer der Anwendung
Die Einheiten von Dysport sind spezifisch für das Präparat Dysport und dürfen nicht auf andere Präparate mit dem Wirkstoff Clostridium botulinum Toxin übertragen werden. Daher wird empfohlen, bei Langzeitbehandlung nicht zwischen unterschiedlichen Clostridium botulinum Toxin-Präparaten zu wechseln.
Dysport darf nur von Ärzten angewendet werden, die in der Behandlung mit Clostridium botulinum Toxin Typ A in der jeweiligen Indikation Erfahrungen besitzen und die erforderliche Ausstattung zur Verfügung haben.
Dysport darf nach Rekonstitution nur für eine Behandlung pro Patient verwendet werden.
Auflösungsvorschrift
Das Herstellen der gebrauchsfertigen Dysport-Injektionslösung erfolgt direkt nach Entnahme aus dem Kühlschrank (siehe Abschnitt 6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung).
Dysport wird mit Hilfe einer Spritze gemäß nachfolgender Tabelle aufgelöst. Es entsteht eine klare Lösung.
|
Durchstechflasche mit 300 Einheiten |
Durchstechflasche mit 500 Einheiten | |
|
Erhaltene Dosis (in Einheiten Clostridium botulinum Toxin Typ A pro 1 ml) |
Zugegebene Menge Lösungsmittel (Natriumchlorid-Inj ektionslösung 9 mg/ml (0,9 %)) |
Zugegebene Menge Lösungsmittel (Natriumchlorid-Inj ektionslösung 9 mg/ml (0,9 %)) |
|
200 Einheiten |
1,5 ml |
2,5 ml |
Hinweis:
Wenn verschiedene Wirkstärken von Dysport während einer Behandlungssitzung angewendet werden, muss darauf geachtet werden, die korrekte Menge an Lösungsmittel zur Auflösung der 200 Einheiten pro 1 ml zu verwenden. Die zuzugebende Menge isotoner Natriumchloridlösung ist für Dysport 300 Einheiten und Dysport 500 Einheiten verschieden. Jede Spritze ist entsprechend zu kennzeichnen.
Blepharospasmus, hemifazialer Spasmus und koexistierende fokale Dystonien
Dosierung
Die empfohlenen Dosierungen gelten für Erwachsene aller Altersgruppen einschließlich älterer Patienten.
Sichere und wirksame Dosierungen von Dysport bei der Behandlung von Blepharospasmus, hemifazialem Spasmus und koexistierenden fokalen Dystonien sind bei Kindern noch nicht ausreichend untersucht.
Bilateraler Blepharospasmus
Bei Behandlungsbeginn insgesamt 40 Einheiten Clostridium botulinum Toxin Typ A pro Auge als Injektion unter die Haut (subkutane Injektion).
Falls erforderlich, kann bei Folgeinjektionen die Dosis auf 60 oder 80 oder maximal 120 Einheiten pro Auge erhöht werden. Eine Erhöhung der Dosis kann jedoch das Risiko für lokale Nebenwirkungen, insbesondere Ptosis, erhöhen.
Die Maximaldosis darf 120 Einheiten pro Auge nicht überschreiten.
Unilateraler Blepharospasmus
Wie bei bilateralem Blepharospasmus. Die Injektion wird jedoch auf das betroffene Auge beschränkt. Hemifazialer Spasmus und koexistierende fokale Dystonien
Wie bei bilateralem Blepharospasmus. Die Injektion wird jedoch auf das betroffene Auge beschränkt. Art der Anwendung
Subkutane Injektion von 10 Einheiten medial und 10 Einheiten lateral in die Verbindung zwischen präseptalem und orbitalem Teil sowohl des oberen (siehe nachfolgende Abbildung, Nummer 1 und 2) als auch des unteren (Nummer 3 und 4) M. orbicularis oculi.
Um das Risiko einer Ptosis zu reduzieren, sollten Injektionen in der Nähe des Levators palpebrae superioris vermieden werden, weshalb bei Injektionen in das obere Lid die Kanüle vom Zentrum weg zu richten ist.
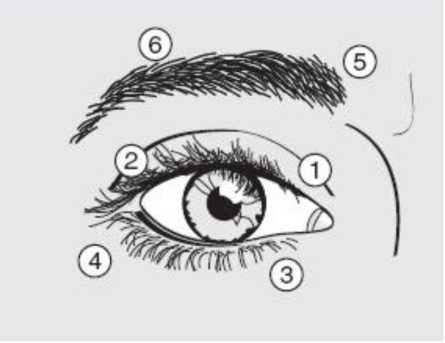
Wenn die Anfangsdosis als nicht ausreichend bewertet wird, kann es bei Folgeinjektionen erforderlich sein, die Dosis pro Auge zu erhöhen auf:
• 60 Einheiten: Es werden jeweils 10 Einheiten medial und 20 Einheiten lateral injiziert.
• 80 Einheiten: Es werden jeweils 20 Einheiten medial und 20 Einheiten lateral injiziert.
• Maximal 120 Einheiten: Es werden jeweils 20 Einheiten medial und 40 Einheiten lateral injiziert.
Zusätzliche Injektionen können in den M. frontalis über der Augenbraue (siehe Abbildung, Nummer 5 und 6) erfolgen, wenn dort befindliche Spasmen das Sehvermögen beeinträchtigen.
Dauer der Anwendung
Eine Besserung der Symptome kann nach 2-4 Tagen, der maximale therapeutische Effekt innerhalb von 2 Wochen erwartet werden.
Die Injektionen sollten ungefähr alle 12 Wochen wiederholt werden oder wann erforderlich, um der Rückkehr der Symptome vorzubeugen, jedoch nicht häufiger als alle 12 Wochen.
Tritt nach Verabreichung der Höchstdosis keine Wirkung ein, so ist der Patient als Therapieversager anzusehen und die Behandlung ist zu beenden.
Zervikale Dystonie (Torticollis spasmodicus)
Dosierung
Insgesamt 500 Einheiten Clostridium botulinum Toxin Typ A in die Hals- und Nackenmuskulatur als streng intramuskuläre Injektion.
Bei wiederholten Injektionen kann es erforderlich sein, die Dosis je nach Ansprechen anzupassen, d. h. entsprechend dem klinischen Zustand um 100-250 Einheiten pro Sitzung schrittweise zu verringern bzw. zu erhöhen, wobei die Maximaldosis von 1.000 Einheiten nicht überschritten werden darf.
Eine Erhöhung der Dosis kann das Risiko für Nebenwirkungen, insbesondere Dysphagie, erhöhen.
Die empfohlenen Dosierungen gelten nur für normalgewichtige Erwachsene, die keine Anzeichen einer verminderten Nackenmuskulatur zeigen. Bei untergewichtigen Patienten und älteren Patienten mit möglicher verminderter Nackenmuskulatur sollte die Dosis reduziert werden.
Sichere und wirksame Dosierungen von Dysport bei der Behandlung des Torticollis spasmodicus bei Kindern sind noch nicht ausreichend untersucht.
Art der Anwendung
Streng intramuskuläre Injektion üblicherweise in den M. sternocleidomastoideus, M. levator scapulae, M. scalenus, M. splenius capitis und/oder M. trapezius.
In den M. sternocleidomastoideus dürfen maximal 300 Einheiten injiziert werden.
Die Identifizierung der Muskeln, in die Dysport injiziert werden soll, richtet sich nach den klinischen Merkmalen (abnorme sichtbare Muskelaktivität, tastbare Verhärtungen, Lokalisierung der Muskelschmerzen) und nach der Verteilung des dystonen EMG-Musters.
Dauer der Anwendung
Eine Besserung der Symptome kann innerhalb 1 Woche erwartet werden.
Die Injektionen sollten ungefähr alle 16 Wochen oder wenn erforderlich bei Rückkehr der Symptome wiederholt werden, jedoch nicht häufiger als alle 12 Wochen.
Tritt nach Verabreichung der Höchstdosis keine Wirkung ein, so ist der Patient als Therapieversager anzusehen und die Behandlung ist zu beenden.
Armspastik bei Erwachsenen infolge eines Schlaganfalls
Dosierung
Insgesamt 1.000 Einheiten Clostridium botulinum Toxin Typ A in die Armmuskulatur als intramuskuläre Injektion. Die Maximaldosis darf 1.000 Einheiten nicht überschreiten.
Die Anfangsdosierung sollte reduziert werden, wenn es Anhaltspunkte für eine übermäßige Schwächung der Zielmuskeln gibt (z. B. bei Patienten mit kleinen Zielmuskeln) oder wenn keine Injektion in den Musculus biceps brachii (BB) erfolgen soll oder wenn gleichzeitig Injektionen anderer Muskelgruppen nötig sind.
Sichere und wirksame Dosierungen von Dysport bei der Behandlung der Armspastik nach Schlaganfall bei Kindern sind noch nicht ausreichend untersucht.
Art der Anwendung
Für die intramuskuläre Injektion werden folgende Injektionsorte und Dosierungen beispielhaft empfohlen:
Dosis (Dysport-Einheiten)
300-400
150
150-250
150
150
max. 1.000
Muskel
M. biceps brachii (BB)
M. flexor digitorum profundus (FDP) M. flexor digitorum superficialis (FDS) M. flexor carpi ulnaris (FCU)
M. flexor carpi radialis (FCR) Gesamtdosis
Es können weitere Muskeln beteiligt sein, die möglicherweise auch behandelt werden müssen.
In den Musculus biceps brachii wird an zwei Stellen injiziert, in alle anderen Muskeln an einer Stelle.
Die Injektionsstellen sollten anhand der Standardstellen für die Elektromyographie sowie durch Palpation festgelegt werden.
Dauer der Anwendung
Eine Besserung der Symptome kann nach zwei Wochen erwartet werden.
Die Injektionen sollten ungefähr alle 16 Wochen oder wenn erforderlich bei Rückkehr der Symptome wiederholt werden, jedoch nicht häufiger als alle 12 Wochen.
4.3 Gegenanzeigen
Dysport darf nicht angewendet werden bei
• nachgewiesener Überempfindlichkeit gegen Clostridium botulinum Toxin Typ A oder einen der sonstigen Bestandteile.
• Infektionen an der Injektionsstelle.
4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Über Nebenwirkungen, für die eine sich von der Injektionsstelle ausbreitende Toxinwirkung verantwortlich gemacht wird, wurde berichtet (siehe Abschnitt 4.8 Nebenwirkungen). Patienten, die mit therapeutischen Dosen behandelt werden, können möglicherweise mit einer übermäßigen Schwächung der Muskulatur reagieren. Das Risiko für das Auftreten solcher Nebenwirkungen könnte reduziert werden, indem die minimal wirksame Dosis angewendet und die empfohlene Dosis nicht überschritten wird.
Nach Behandlung mit Clostridium botulinum Toxin Typ A oder B wurde sehr selten von Todesfällen berichtet, die vereinzelt mit Dysphagie und/oder Aspirationspneumonie (unter anderem Dyspnoe, Lungeninsuffizienz, Atemstillstand) assoziiert waren und/oder bei Patienten mit bedeutsamer Asthenie auftraten.
Bei Patienten mit Erkrankungen wie gestörter neuromuskulärer Übertragung, Schluck- und Atemschwierigkeiten besteht ein erhöhtes Risiko für das Auftreten solcher Wirkungen. Bei diesen Patienten muss die Behandlung unter fachärztlicher Kontrolle erfolgen und nur dann, wenn der Nutzen der Behandlung das Risiko überwiegt.
Bei Patienten mit bestehenden Schluck- und Atemschwierigkeiten sollte Dysport mit Vorsicht angewendet werden, weil diese sich verstärken können, falls sich die Toxinwirkung zu den betreffenden Muskeln ausbreitet. Aspiration trat in seltenen Fällen auf und ist ein Risiko, wenn Patienten behandelt werden, die eine chronische Atemstörung haben.
Bei Patienten mit subklinischem oder klinischem Befund einer merklich gestörten neuromuskulären Übertragung (z. B. Myasthenia gravis) sollte Dysport nur mit Vorsicht und unter enger Überwachung angewendet werden. Diese Patienten können mit einer erhöhten Empfindlichkeit auf Substanzen wie Dysport reagieren, was zu einer übermäßigen Schwächung der Muskulatur führen kann.
Die empfohlene Dosierung und Häufigkeit der Anwendung von Dysport darf nicht überschritten werden (siehe Abschnitt 4.2 Dosierung, Art und Dauer der Anwendung).
Der Patient und pflegende Personen sind darauf hinzuweisen, dass der ärztliche Notdienst sofort zu benachrichtigen ist, wenn Schluck-, Sprech- bzw. Atemstörungen auftreten.
Dysport sollte bei Patienten, die eine fixe Kontraktur entwickelt haben, nicht angewendet werden.
Wechselhaftes klinisches Ansprechen, wie es bei Folgeinjektionen mit Dysport (und allen anderen Botulinumtoxinen) auftreten kann, ist möglicherweise auf unterschiedliches Vorgehen beim
Rekonstituieren, die gewählten Injektionsintervalle, die injizierten Muskeln und eine geringfügig variierende Aktivität des Toxins, bedingt durch die verwendete biologische Testmethode, zurückzuführen.
Wie bei jeder intramuskulären Injektion, sollte Dysport bei Patienten mit Blutgerinnungsstörungen oder Infektionen bzw. Entzündungen an der geplanten Injektionsstelle nur angewendet werden, wenn dies eindeutig erforderlich ist.
Eine Durchstechflasche Dysport darf nur bei einem einzigen Patienten, während einer einzigen Behandlung angewendet werden. Nicht verwendetes Dysport muss, wie in Abschnitt 6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung beschrieben, entsorgt werden. Bestimmte Vorsichtsmaßnahmen müssen bei der Zubereitung und Verabreichung des Produkts beachtet werden, wie auch bei der Inaktivierung und Entsorgung von nicht verwendeter Injektionslösung (siehe Abschnitt 6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung).
Dysport enthält eine geringe Menge Humanalbumin. Das Risiko einer Übertragung von viralen Infektionen kann nach Verwendung von menschlichem Blut oder Blutprodukten nicht mit absoluter Sicherheit ausgeschlossen werden.
Antikörperbildung gegenüber Clostridium botulinum Toxin ist bei mit Dysport behandelten Patienten selten beobachtet worden. Klinisch können neutralisierende Antikörper durch eine deutliche Verminderung des Therapieerfolges und/oder die Notwendigkeit stetiger Dosiserhöhungen vermutet werden. Da das Risiko der Antikörperbildung bei höheren Dosen und kurzen Dosierungsintervallen ansteigt, sollte die minimal wirksame Dosis innerhalb größtmöglicher Therapieintervalle angewendet werden.
4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Die Wirkung von Clostridium botulinum Toxin kann durch Arzneimittel, die direkt oder indirekt die neuromuskuläre Übertragung beeinträchtigen (z. B. Aminoglykoside; Curare-artige, nicht depolarisierende Blocker), theoretisch verstärkt werden. Solche Arzneimittel sollten bei mit Clostridium botulinum Toxin behandelten Patienten mit Vorsicht angewendet werden.
4.6 Schwangerschaft und Stillzeit
Es gibt nur wenige Daten zur Anwendung von Clostridium botulinum Toxin Typ A beim Menschen während der Schwangerschaft. Abgesehen von hohen Dosen, die mütterliche Toxizität bewirken, lassen tierexperimentelle Studien nicht auf direkte oder indirekte schädliche Auswirkungen auf Schwangerschaft, embryonale/fetale Entwicklung, Geburt oder postnatale Entwicklung schließen (siehe Abschnitt 5.3).
Dysport darf während der Schwangerschaft nur angewendet werden, wenn es eindeutig erforderlich ist. Bei der Anwendung in der Schwangerschaft ist Vorsicht geboten.
Es ist nicht bekannt, ob Clostridium botulinum Toxin Typ A in die Muttermilch übergeht.
Die Anwendung von Clostridium botulinum Toxin Typ A während der Stillzeit kann daher nicht empfohlen werden.
4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Schwächung der Muskulatur und Ermüdung oder Sehstörungen sind mögliche Risiken, die, sofern sie auftreten, vorübergehend die Verkehrstüchtigkeit oder die Fähigkeit zum Bedienen von Maschinen beeinträchtigen können.
4.8 Nebenwirkungen
Nebenwirkungen können aufgrund einer zu tief oder falsch platzierten Injektion von Dysport, die zu einer vorübergehenden Paralyse nahe liegender Muskelgruppen führen kann, auftreten.
Bei den Häufigkeitsangaben zu Nebenwirkungen werden folgende Kategorien zugrunde gelegt:
Sehr häufig (> 1/10); häufig (> 1/100, < 1/10); gelegentlich (> 1/1.000, < 1/100); selten (> 1/10.000,
< 1/1.000); sehr selten (< 1/10.000).
Alle Indikationen
Während klinischer Studien trat bei ungefähr 25 % der mit Dysport behandelten Patienten eine unerwünschte Wirkung auf. Die folgenden Nebenwirkungen wurden bei Patienten beobachtet, die aufgrund verschiedener Indikationen, einschließlich der nachfolgend im Einzelnen aufgeführten, behandelt wurden:
Erkrankungen des Nervensystems:
Selten: Neuralgische Muskelatrophien.
Erkrankungen der Haut und des Unterhautzellgewebes:
Gelegentlich: Juckreiz.
Selten: Hautrötung.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort:
Häufig: Allgemeines Schwächegefühl, Ermüdung, grippeähnliche Symptome, Hämatome und/oder Schmerzen an der Injektionsstelle.
Zusätzlich wurde über folgende indikationsspezifische Nebenwirkungen berichtet:
Blepharospasmus, hemifazialer Spasmus und koexistierende fokale Dystonien
Erkrankungen des Nervensystems:
Häufig: Schwächung der Gesichtsmuskulatur.
Gelegentlich: Fazialisparese (Gesichtslähmung).
Augenerkrankungen:
Sehr häufig: Ptosis.
Häufig: Diplopie, Augentrockenheit, tränende Augen.
Selten: Ophthalmoplegie.
Erkrankungen der Haut und des Unterhautzellgewebes:
Häufig: Schwellung des Augenlids.
Selten: Entropium.
Zervikale Dystonie (Torticollis spasmodicus)
Erkrankungen des Nervensystems:
Häufig: Kopfschmerzen, Schwindel, Fazialisparese (milde und unvollständige Gesichtslähmung). Augenerkrankungen:
Häufig: Verschwommensehen, reduzierte Sehschärfe.
Gelegentlich: Diplopie, Ptosis.
Erkrankungen der Atemwege, des Brustraums und Mediastinums:
Häufig: Dysphonie, Dyspnoe.
Selten: Aspiration.
Erkrankungen des Gastrointestinaltrakts:
Sehr häufig: Dysphagie, Mundtrockenheit.
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen:
Sehr häufig: Muskelschwäche.
Häufig: Nackenschmerzen, Schmerzen der Skelettmuskulatur, Myalgie, Schmerzen in den Extremitäten, Versteifung der Skelettmuskulatur.
Gelegentlich: Muskelatrophie, Kieferfunktionsstörung.
Die Dysphagie schien dosisabhängig zu sein und trat am häufigsten nach Injektion in den M. sternocleidomastoideus auf. Weiche Nahrung kann erforderlich sein bis die Symptome abklingen.
Armspastik bei Erwachsenen infolge eines Schlaganfalls
Erkrankungen des Gastrointestinaltrakts:
Häufig: Dysphagie.
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen:
Häufig: Schwächung der Armmuskulatur.
Verletzung, Vergiftung und durch Eingriffe bedingte Komplikationen:
Häufig: Zufällige Verletzungen und Stürze.
Erfahrungen zur Sicherheit nach Markteinführung
Das Nebenwirkungsprofil, das dem Zulassungsinhaber seit Markteinführung übermittelt wurde, spiegelt die Pharmakologie des Produkts wider und entspricht dem während klinischer Studien beobachteten.
Es gab vereinzelt Berichte über Überempfindlichkeitsreaktionen (einschließlich Urtikaria, Angioödem, Pharyngealödem, Atemprobleme).
Über Nebenwirkungen, für die eine sich von der Injektionsstelle ausbreitende Toxinwirkung verantwortlich gemacht wird (übermäßige Schwächung der Muskulatur, Dysphagie, Aspirationspneumonie, die tödlich sein können), wurde sehr selten berichtet.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Arzneimittel und Medizinprodukte, Abt. Pharmakovigilanz, Kurt-Georg-Kiesinger Allee 3, D-53175 Bonn, Website: http://www.bfarm.de anzuzeigen.
4.9 Überdosierung
Exzessive Dosierungen können von der Injektionsstelle entfernte und ausgeprägte neuromuskuläre Lähmungen erzeugen. Bei Überdosierungen besteht ein erhöhtes Risiko, dass das Neurotoxin in die Blutbahn gelangt und zu Komplikationen führt, wie sie nach oralen Botulinum-Intoxikationen auftreten (z. B. Dysphagie und Dysphonie). Künstliche Beatmung kann erforderlich sein, wenn exzessive Dosen die Atemmuskeln lähmen. Es gibt kein spezifisches Antidot; vom Antitoxin ist keine therapeutische Wirkung zu erwarten. Es werden allgemeine unterstützende Maßnahmen empfohlen.
Im Fall einer Überdosierung sollte der Patient auf Symptome übermäßiger Muskelschwäche oder Muskelparalyse medizinisch überwacht werden. Eine symptomatische Behandlung sollte, falls notwendig, durchgeführt werden.
Symptome der Überdosierung treten möglicherweise nicht sofort nach Injektion auf. Bei versehentlicher Überdosierung oder oraler Aufnahme sollte die Person über mehrere Wochen auf Anzeichen und Symptome übermäßiger Muskelschwäche oder Muskelparalyse medizinisch überwacht werden.
5. PHARMAKOLOGISCHE EIGENSCHAFTEN
5.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Andere peripher wirkende Muskelrelaxanzien ATC-Code: M03AX21
Clostridium botulinum Toxin Typ A blockiert die cholinerge Übertragung an der motorischen Endplatte durch eine Unterbindung der Freisetzung von Acetylcholin. Der Wirkungsmechanismus des Neurotoxins umfasst zunächst eine spezifische und sättigbare Bindung an die extrazellulären Rezeptoren. Durch einen pinozytoseähnlichen, energieabhängigen Prozess wird das Neurotoxin dann ins Zellinnere (Internalisation des Toxins) aufgenommen. Im Zellinnern wirkt das Neurotoxin wahrscheinlich als Enzym auf eine zelluläre Komponente ein, deren Funktion in der Regulierung der Calcium-abhängigen Exozytose von Acetylcholin besteht. Die Nervenendigungen der motorischen Endplatte sprechen auf die Nervenimpulse nicht mehr an und es erfolgt keine Sekretion des Chemotransmitters (chemische Denervierung des Muskels). Nach Eindringen des Neurotoxins ins Zellinnere erfolgt wahrscheinlich eine enzymatische Zersetzung der Strukturen der motorischen Endplatte.
Die Wiederherstellung der Impulsübertragung erfolgt über die neu gebildeten Nervenendigungen und motorischen Endplatten, die während der Regenerationsprozesse entstehen. Der Prozess der Neubildung von Nervenendigungen beginnt ca. 8 Wochen nach Toxinverabreichung. Diese Strukturen weichen anatomisch teilweise von den bisherigen ab, sind jedoch ausreichend funktionsfähig. Nach wiederholter Verabreichung des Neurotoxins werden die neu gebildeten Nervenendigungen und motorischen Endplatten erneut lädiert. Die Langzeitfolgen dieser kumulativen Schädigung sind nicht bekannt.
Tierexperimentelle Untersuchungen zeigen, dass Clostridium botulinum Toxin Typ A die cholinerge sympathische und parasympathische Impulsübertragung blockieren kann. Die Impulsleitung entlang der Nerven wird wahrscheinlich nicht beeinflusst. Das Neurotoxin hat keinen Einfluss auf die Funktion sensibler Nervenendigungen.
In vitro-Untersuchungen zeigen, dass Clostridium botulinum Toxin Typ A die Freisetzung anderer Neurotransmitter als Acetylcholin hemmen kann. Unbeeinflusst bleibt lediglich die purinerge und peptiderge Impulsübertragung.
5.2 Pharmakokinetische Eigenschaften
Die pharmakologischen Effekte, die nach oraler und parenteraler Verabreichung von Clostridium botulinum Toxin Typ A beim Menschen beobachtet werden, zeigen, dass das Neurotoxin aus dem Gastrointestinaltrakt und Muskelgewebe umfangreich absorbiert wird.
Die hinreichenden pharmakokinetischen Untersuchungen am Tier sind dadurch erschwert, dass das Neurotoxin eine hohe Wirkpotenz entwickelt, die anzuwendenden Dosen sehr klein sind und die
Markierung des Neurotoxins zur Erzeugung einer ausreichend hohen spezifischen Aktivität sehr schwer ist.
Untersuchungen zu Dosis/Zeit/Wirkungsbeziehungen an Affen zeigten, dass die Wirkung bei niedriger Dosierung mit einer Latenz von 2 bis 3 Tagen nach Injektion auftrat. Die Wirkdauer schwankte zwischen 2 Wochen und 8 Monaten. Ähnliche Dosis/Zeit/Wirkungsbeziehungen wurden auch am Menschen beobachtet.
Das Neurotoxin diffundiert von der Injektionsstelle in das benachbarte Gewebe. Der Umfang der Diffusion ist von den anatomischen Gegebenheiten (Aponeurosen und Faszien stellen ein mechanisches Hindernis dar), vom Injektionsvolumen und von der Dosis abhängig (je größer das Injektionsvolumen und je höher die Dosis, desto ausgeprägter und häufiger sind die unerwünschten Wirkungen, die aus der Toxinverbreitung resultieren).
Das Neurotoxin, das in die Nervenfortsätze der motorischen Nervenzellen aufgenommen wird, wird intraaxonal retrograd transportiert. Radioaktiv markiertes Material wurde im Soma der Motoneurone im Rückenmark nachgewiesen, aber ohne funktionelle Aktivität des Materials. Dagegen konnte das Neurotoxin nicht in den sensiblen Nervenfasern gefunden werden.
Autoradiographische Untersuchungen unter Anwendung des I markierten Clostridium botulinum Toxin Typ A zeigten, dass die Radioaktivität nach Verabreichung niedrigerer Dosen des Neurotoxins auf die motorischen Endplatten an bzw. in der Nähe der Injektionsstelle beschränkt ist. Höhere Dosen führten zur breiteren Verteilung des Neurotoxins mit darauf folgender Paralyse von Muskeln, die von der Injektionsstelle entfernt lagen.
Untersuchungen zum Metabolismus und zur Ausscheidung des Neurotoxins liegen nicht vor. Es wird angenommen, dass das Neurotoxin möglicherweise durch extra- und intrazelluläre Proteolyse abgebaut wird. Es wird vermutet, dass die durch Proteolyse im Zellinnern entstandenen Aminosäuren in den eigenen Aminosäuren-Pool aufgenommen werden könnten.
5.3 Präklinische Daten zur Sicherheit
Eine chronische Toxizitätsstudie, die an Ratten mit bis zu 12 Einheiten/Tier durchgeführt wurde, ergab keine Anzeichen für eine systemische Toxizität. Effekte in Tierstudien zur Reproduktions- und chronischen Toxizität waren überwiegend auf die mit der Wirkung von Clostridium botulinum Toxin Typ A zusammenhängenden Änderungen am injizierten Muskel begrenzt. An Kaninchenaugen wurde nach Anwendung von Clostridium botulinum Toxin Typ A keine Reizung beobachtet.
Studien zur Reproduktionstoxizität an trächtigen Ratten und Kaninchen, bei denen jeweils täglich Clostridium botulinum Toxin Typ A in Dosen bis zu 79 Einheiten/kg und 42 Einheiten/kg intramuskulär angewendet wurde, ergaben keine embryonale/fetale Toxizität. Bei höheren Dosen trat bei beiden Spezies eine schwere mütterliche Toxizität auf, die mit ausbleibender embryonaler Einnistung verbunden war. Clostridium botulinum Toxin Typ A zeigte weder bei Ratten noch bei Kaninchen teratogene Eigenschaften; auch in prä- und postnatalen Studien wurden bei Ratten in der F1-Generation keine Effekte beobachtet. Die Fertilität war bei männlichen und weiblichen Tieren aufgrund eingeschränkter Paarung vermindert, was bei hohen Dosen auf die Muskelparalyse zurückzuführen ist.
6. PHARMAZEUTISCHE ANGABEN
6.1 Liste der sonstigen Bestandteile
Humanalbumin und Lactose-Monohydrat.
6.2 Inkompatibilitäten
Das Arzneimittel darf, außer mit den unter Abschnitt 4.2 Auflösungsvorschrift aufgeführten, nicht mit anderen Arzneimitteln gemischt werden. Es sind keine Kompatibilitätsstudien mit anderen Arzneimitteln, außer den unter Abschnitt 4.2 Auflösungsvorschrift genannten, durchgeführt worden.
6.3 Dauer der Haltbarkeit
Haltbarkeit des Pulvers im unversehrten Behältnis: 2 Jahre.
Haltbarkeit nach Herstellung der gebrauchsfertigen Injektionslösung:
Nach Rekonstitution wurde die Haltbarkeit bei 2°C - 8°C für 24 Stunden nachgewiesen.
Aus mikrobiologischer Sicht sollte die gebrauchsfertige Injektionslösung sofort angewendet werden. Wenn sie nicht sofort angewendet wird, ist der Anwender für die Dauer und die Bedingungen der Lagerung verantwortlich, die nicht länger als 24 Stunden bei 2°C - 8°C betragen sollte.
6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Im Kühlschrank lagern (2°C - 8°C).
Gebrauchsfertige Injektionslösung bei 2°C - 8°C maximal 24 Stunden aufbewahren.
6.5 Art und Inhalt des Behältnisses
Durchstechflasche (Glas Typ I) mit einem Butylgummistopfen und einer Aluminium-Bördelkappe mit Plastikabdeckung.
Dysport 300 Einheiten: Originalpackung mit 1, 2 oder 6 Durchstechflaschen.
Dysport 500 Einheiten: Originalpackung mit 1, 2, 3 oder 6 Durchstechflaschen, Klinikpackung mit 6 oder 12 Durchstechflaschen.
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung
Handhabung
Siehe Abschnitt 4.2 Auflösungsvorschrift.
Beseitigung
Clostridium botulinum Toxin ist sehr empfindlich gegenüber Hitze und Chemikalien.
Verschüttetes Dysport-Pulver muss mit einem saugfähigen Tuch, das mit verdünnter HypochloritLösung (1 % freies Chlor) getränkt wurde, aufgewischt werden. Verschüttete Dysport-Injektionslösung muss mit einem trockenen, saugfähigen Tuch aufgewischt werden.
Die verunreinigten Oberflächen sollten mit einem saugfähigen Tuch, das mit verdünnter HypochloritLösung (1 % freies Chlor) getränkt wurde, gereinigt und anschließend trockengerieben werden.
Falls eine Durchstechflasche zerbrochen ist, sollten die Glasscherben vorsichtig gesammelt und das Pulver bzw. die Flüssigkeit wie oben angegeben aufgewischt werden, wobei Hautverletzungen vermieden werden müssen.
Nach Hautkontakt mit dem Produkt muss die betroffene Hautfläche mit reichlich Wasser gewaschen werden.
Nach Augenkontakt mit dem Produkt muss das betroffene Auge 15 Minuten mit viel Wasser oder steriler isotoner Natriumchloridlösung gespült werden.
Im Fall einer Verletzung des Anwenders (durch Schnitt oder Injektion) muss die betroffene Hautfläche mit reichlich Wasser gewaschen werden. Abhängig von der injizierten Dosis sollten entsprechende medizinische Maßnahmen ergriffen werden.
Empfehlungen für die Beseitigung der kontaminierten Gegenstände
Injektionsnadeln, Injektionsspritzen und Durchstechflaschen - die nicht geleert werden sollten -müssen in einen geeigneten Behälter, der nach Gebrauch der Müllverbrennung zugeführt wird, entsorgt werden.
Kontaminierte Materialien (saugfähige Tücher, Handschuhe, Glasscherben) sollten in einem für spitze Gegenstände geeigneten Behälter der Müllverbrennung zugeführt werden.
7. INHABER DER ZULASSUNG
Ipsen Pharma GmbH 76275 Ettlingen Tel.: 07243 184-80 Fax: 07243 184-39
8. ZULASSUNGSNUMMER
Dysport 300 Einheiten: 81122.00.00 Dysport 500 Einheiten: 50586.00.00
9. DATUM DER ERTEILUNG DER ZULASSUNG
Dysport 300 Einheiten: 03. April 2012 Dysport 500 Einheiten: 04. Juni 2002
10. STAND DER INFORMATION
Januar 2014
11. VERKAUFSABGRENZUNG
Verschreibungspflichtig
spcde-dys300-500u-gl03clean.rtf Seite 12 von 12