Eligard 7,5Mg
ZUSAMMENFASSUNG DER MERKMALE DES ARZNEIMITTELS
1. BEZEICHNUNG DES ARZNEIMITTELS
ELIGARD 7,5 mg Pulver und Lösungsmittel zur Herstellung einer Injektionslösung
2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
1 vorgefüllte Spritze mit Pulver zur Herstellung einer Injektionslösung enthält 7,5 mg Leuprorelinacetat, entsprechend 6,96 mg Leuprorelin.
Vollständige Auflistung der sonstigen Bestandteile siehe, Abschnitt 6.1.
3. DARREICHUNGSFORM
Pulver und Lösungsmittel zur Herstellung einer Injektionslösung Pulver (Spritze B):
vorgefüllte Spritze mit weißem bis gebrochen weißem Pulver Lösungsmittel (Spritze A):
vorgefüllte Spritze mit klarer, farbloser bis blassgelber Lösung
4. KLINISCHE ANGABEN
4.1 Anwendungsgebiete
ELIGARD 7,5 mg ist für die Behandlung des hormonabhängigen, fortgeschrittenen Prostatakarzinoms und in Kombination mit Radiotherapie für die Behandlung von lokalisiertem Hochrisiko- und lokal fortgeschrittenem hormonabhängigem Prostatakarzinom indiziert.
4.2 Dosierung und Art der Anwendung
Dosierung
Erwachsene Männer
ELIGARD 7,5 mg sollte unter Anleitung eines Mediziners verabreicht werden, der über eine angemessene Erfahrung in der Kontrolle des Ansprechens auf die Therapie verfügt.
ELIGARD 7,5 mg wird einmal monatlich als subkutane Injektion verabreicht. Die injizierte Lösung bildet ein festes Arzneimitteldepot und ermöglicht einen Monat lang eine kontinuierliche Freisetzung von Leuprorelinacetat.
In der Regel erfordert die Therapie des fortgeschrittenen Prostatakarzinoms mit ELIGARD 7,5 mg eine langfristige Behandlung, die nicht abgebrochen werden sollte, wenn eine Remission oder Besserung eintritt.
ELIGARD 7,5 mg kann als neoadjuvante oder adjuvante Therapie in Kombination mit Radiotherapie bei lokalisiertem Hochrisiko- und lokal fortgeschrittenem Prostatakarzinom angewendet werden.
Das Ansprechen auf ELIGARD 7,5 mg sollte mittels klinischer Parameter und durch Bestimmung des prostataspezifischen Antigens (PSA) im Serum überwacht werden. Klinische Studien haben gezeigt, dass die Testosteronspiegel im Serum in den ersten 3 Behandlungstagen bei den meisten nicht orchi-ektomierten Patienten ansteigen und dann innerhalb von 3 bis 4 Wochen unter die Kastrationsschwelle absinken. Nach Erreichen des Kastrationsbereichs bleiben die Testosteronspiegel in diesem Bereich, solange die Arzneitherapie fortgesetzt wird (Testosteron-Durchbruchsphänomen < 1 %). Bei einem anscheinend suboptimalen Ansprechen des Patienten sollte überprüft werden, ob der
Testosteronspiegel im Serum den Kastrationsbereich erreicht hat oder auf diesem Niveau bleibt. Da aus fehlerhafter Zubereitung, Rekonstitution oder Verabreichung mangelnde Wirksamkeit resultieren kann, sollten die Testosteronspiegel kontrolliert werden, wenn Fehler bei der Handhabung vermutet werden oder bekannt sind (siehe Abschnitt 4.4).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit wurde bei Kindern im Alter von 0 bis 18 Jahren nicht untersucht (siehe auch Abschnitt 4.3).
Spezielle Patientengruppen
Es wurden keine klinischen Studien bei Patienten mit eingeschränkter Leber- oder Nierenfunktion durchgeführt.
Art der Anwendung
ELIGARD 7,5 mg darf nur von medizinischem Fachpersonal zubereitet, rekonstituiert oder verabreicht werden, das mit der sachgemäßen Handhabung vertraut ist. Siehe Abschnitt 6.6: Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung. Falls das Produkt nicht korrekt zubereitet wurde, darf es nicht verabreicht werden.
Der Inhalt der beiden vorgefüllten sterilen Spritzen ist unmittelbar vor der subkutanen Injektion von ELIGARD 7,5 mg zu mischen.
Eine intraarterielle oder intravenöse Injektion ist aufgrund tierexperimenteller Befunde unbedingt zu vermeiden.
Wie bei anderen subkutan verabreichten Arzneimitteln sollte die Injektionsstelle regelmäßig gewechselt werden.
4.3 Gegenanzeigen
ELIGARD 7,5 mg ist bei Frauen und Kindern kontraindiziert.
Überempfindlichkeit gegen Leuprorelinacetat, andere GnRH-Agonisten oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Orchiektomierte Patienten (wie bei anderen GnRH-Agonisten führt ELIGARD 7,5 mg im Falle einer chirurgischen Kastration nicht zu einem weiteren Absinken des Serumtestosterons).
Als alleinige Behandlung bei Prostatakarzinom-Patienten mit Rückenmarkkompression oder Anzeichen von Metastasen im Rückenmark (siehe auch Abschnitt 4.4).
4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Mangelnde klinische Wirksamkeit kann aufgrund fehlerhafter Rekonstitution des Produktes auftreten. Siehe Abschnitt 4.2 und Abschnitt 6.6 zu den Anweisungen für die Zubereitung und die Verabreichung des Produktes sowie für die Kontrolle der Testosteronspiegel, wenn Fehler bei der Handhabung vermutet werden oder bekannt sind.
Eine Androgendeprivationstherapie kann das QT-Intervall verlängern.
Bei Patienten mit einer Vorgeschichte einer QT-Verlängerung oder mit Risikofaktoren für eine QT-Verlängerung und bei Patienten, die als Begleitmedikation Arzneimittel erhalten, die das QT-Intervall verlängern können (siehe Abschnitt 4.5), sollten die Ärzte das Nutzen-Risiken-Verhältnis einschließlich dem möglichen Auftreten von Torsade de Pointes abwägen, bevor die Behandlung mit ELIGARD 7,5 mg begonnen wird.
Wie andere GnRH-Agonisten führt Leuprorelinacetat in der ersten Behandlungswoche zu einem vorübergehenden Anstieg der Serumkonzentrationen von Testosteron, Dihydrotestosteron und der sauren Phosphatase. Dabei kann es zu einer Verschlechterung der Symptome oder zum Auftreten neuer Symptome kommen, wie Knochenschmerzen, Neuropathie, Hämaturie oder Obstruktion von Ureter oder Blasenausgang (siehe Abschnitt 4.8). Diese Symptome klingen gewöhnlich bei Fortsetzung der Therapie wieder ab.
Die zusätzliche Gabe eines geeigneten Antiandrogens 3 Tage vor Beginn und in den ersten 2 bis 3 Wochen der Leuprorelintherapie sollte daher in Betracht gezogen werden. Berichten zufolge sollen sich die Folgen eines initialen Anstiegs des Serumtestosterons dadurch verhindern lassen.
Nach einer chirurgischen Kastration bewirkt ELIGARD 7,5 mg bei Männern kein weiteres Absinken des Serumtestosteronspiegels.
Über Fälle von Ureterobstruktion und Rückenmarkkompression, die zu Lähmungserscheinungen mit oder ohne letale Komplikationen beitragen können, wurde im Zusammenhang mit GnRH-Agonisten berichtet. Bei Entstehung einer Rückenmarkkompression oder Nierenfunktionsstörung sollte die übliche Behandlung solcher Komplikationen eingeleitet werden.
Patienten mit vertebralen Metastasen und/oder Hirnmetastasen wie auch Patienten mit einer Obstruktion im Bereich des Harntrakts sollten während der ersten Behandlungswochen sorgfältig überwacht werden.
Manche Patienten leiden an Tumoren, die sich durch Hormone nicht beeinflussen lassen. Wenn es trotz entsprechender Testosteronsuppression nicht zu einer klinischen Besserung kommt, ist dies ein Hinweis auf eine Erkrankung, die auf eine Fortsetzung der Behandlung mit ELIGARD 7,5 mg nicht ansprechen wird.
In der Fachliteratur wird über eine Verminderung der Knochendichte bei Männern nach Orchiektomie oder nach Therapie mit einem GnRH-Agonisten berichtet (siehe Abschnitt 4.8).
Unter einer antiandrogenen Therapie besteht ein deutlich erhöhtes Risiko der Entstehung Osteoporosebedingter Frakturen. Hierzu liegen nur in begrenztem Umfang Daten vor. Durch Osteoporose bedingte Frakturen wurden nach 22 Monaten einer pharmakologischen Androgenentzugstherapie bei 5 % der Patienten und nach einer 5 bis 10 Jahre dauernden Behandlung bei 4 % der Patienten beobachtet. Gewöhnlich ist das Risiko einer Osteoporose-bedingten Fraktur höher als das Risiko einer pathologischen Fraktur.
Neben einem lang anhaltenden Testosteronmangel kann das Osteoporoserisiko auch durch hohes Alter, Rauchen, Alkohol, Fettleibigkeit und mangelnde Bewegung beeinflusst werden.
Im Rahmen der Anwendungsbeobachtung wird über seltene Fälle einer Hypophysenapoplexie (klinisches Syndrom infolge eines Hypophyseninfarkts) nach Verabreichung von GnRH-Agonisten berichtet. In den meisten Fällen trat diese Komplikation innerhalb von 2 Wochen nach der ersten Gabe, manchmal schon innerhalb einer Stunde auf. In diesen Fällen ging die Hypophysenapoplexie mit plötzlich auftretenden Kopfschmerzen, Erbrechen, Veränderungen des Sehvermögens, Ophthalmoplegie, verändertem Geisteszustand und zuweilen mit Kreislaufzusammenbruch einher. Solche Fälle bedürfen einer sofortigen ärztlichen Versorgung.
Hyperglykämie und Diabetes: Hyperglykämie und ein erhöhtes Risiko, Diabetes zu entwickeln, wurden bei Männern, die GnRH-Agonisten erhielten, beobachtet. Hyperglykämie könnte ein Zeichen der Entwicklung eines Diabetes mellitus oder der Verschlechterung der glykämischen Kontrolle bei Patienten mit Diabetes sein. Bei Patienten, die GnRH-Agonisten erhalten, sollte der Blutzucker und/oder glykosyliertes Hämoglobin (HbA1c) regelmäßig kontrolliert werden und die Patienten sollten gemäß der gültigen klinischen Praxis behandelt werden.
Kardiovaskuläre Erkrankungen: Ein erhöhtes Risiko für die Entwicklung eines Myokardinfarkts, plötzlichen Herztods und Schlaganfalls wurde in Zusammenhang mit dem Einsatz von GnRH-Agonisten bei Männern berichtet. Das Risiko erscheint, basierend auf den berichteten Odds Ratios, gering und sollte sorgsam zusammen mit den kardiovaskulären Risikofaktoren abgewogen werden, wenn die Behandlung für einen Patienten mit Prostatakarzinom festgelegt wird. Patienten, die GnRH-Agonisten erhalten, sollten auf Anzeichen und Symptome einer Entwicklung einer kardiovaskulären Erkrankung beobachtet und gemäß der gültigen klinischen Praxis behandelt werden.
4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Es wurden keine pharmakokinetischen Wechselwirkungsstudien mit ELIGARD 7,5 mg durchgeführt. Über Wechselwirkungen von Leuprorelinacetat mit anderen Arzneimitteln liegen keine Berichte vor.
Da eine Androgendeprivationstherapie das QT-Intervall verlängern kann, ist die gleichzeitige Anwendung von ELIGARD 7,5 mg und Arzneimitteln, für die bekannt ist, dass sie das QT-Intervall verlängern, oder Arzneimitteln, die Torsade de Pointes hervorrufen können, wie Antiarrhythmika der Klasse IA (z. B. Chinidin, Disopyramid) oder der Klasse III (z. B. Amiodaron, Sotalol, Dofetilid, Ibutilid), Methadon, Moxifloxacin, Antipsychotika etc. sorgfältig abzuwägen (siehe Abschnitt 4.4).
4.6 Fertilität, Schwangerschaft und Stillzeit
Nicht zutreffend (ELIGARD 7,5 mg ist bei Frauen kontraindiziert).
4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Es wurden keine Studien zu den Auswirkungen einer Behandlung mit ELIGARD 7,5 mg auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen durchgeführt.
Die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen kann durch Müdigkeit, Schwindel und Sehstörungen (mögliche Nebenwirkungen der Behandlung oder Folgen einer Grunderkrankung) beeinträchtigt werden.
4.8 Nebenwirkungen
Unerwünschte Reaktionen auf ELIGARD hängen hauptsächlich mit spezifischen pharmakologischen Wirkungen von Leuprorelin zusammen (Anstieg und Absinken bestimmter Hormonspiegel). Am häufigsten wird über Hitzewallungen, Unwohlsein, Übelkeit und Müdigkeit sowie über vorübergehende lokale Reizung an der Injektionsstelle berichtet. Bei ca. 58 % der Patienten kommt es zu leichten bis mittelschweren Hitzewallungen.
Tabellarische Zusammenfassung der Nebenwirkungen
Über die folgenden unerwünschten Ereignisse wurde im Rahmen klinischer Studien mit ELIGARD an Patienten mit fortgeschrittenem Prostatakarzinom berichtet. Bezüglich der Häufigkeit solcher Ereignisse unterscheidet man zwischen sehr häufig (> 1/10), häufig (> 1/100 bis < 1/10), gelegentlich (> 1/1.000 bis < 1/100), selten (> 1/10.000 bis < 1/1.000), sehr selten (< 1/10.000) und nicht bekannt (Häufigkeit auf Grund der verfügbaren Daten nicht abschätzbar).
|
Infektionen und parasitäre Erkrankungen Häufig: Gelegentlich: Stoffwechsel- und Ernährungsstörungen |
Nasopharyngitis Harnwegsinfektion, lokale Infekte der Haut |
|
Gelegentlich: |
Verschlechterung eines Diabetes mellitus |
|
Psychiatrische Erkrankungen Gelegentlich: |
abnorme Träume, Depression, Abnahme der Libido |
|
Erkrankungen des Nervensystems Gelegentlich: Selten: |
Schwindel, Kopfschmerzen, Insomnie, Geschmacks- und Geruchsstörungen, verminderte Reizempfindung abnorme unwillkürliche Bewegungen |
|
Herzerkrankungen Nicht bekannt: |
QT-Verlängerung (siehe Abschnitte 4.4 und 4.5) |
|
Gefäßerkrankungen Sehr häufig: Gelegentlich: Selten: |
Hitzewallungen Hypertonie, Hypotonie Synkope, Kollaps |
|
Erkrankungen der Atemwege, des Brustraums und Mediastinums Gelegentlich: |
Rhinorrhoe, Dyspnoe |
|
Erkrankungen des Gastrointestinaltrakts Häufig: Gelegentlich: Selten: |
Übelkeit, Diarrhö Obstipation, Mundtrockenheit, Erbrechen, Dyspepsie Flatulenz, Aufstoßen |
|
Erkrankungen der Haut und des Unterhautzellgewebes Sehr häufig: Häufig: Gelegentlich: Selten: |
Ekchymose, Erythem Pruritus, Nachtschweiß feuchtkalte Haut, vermehrtes Schwitzen Alopezie, Hautausschlag |
|
Skelettmuskulatur- und Bindegewebserkrankungen Häufig: Gelegentlich: |
Arthralgie, Schmerzen in den Extremitäten, Myalgie Rückenschmerzen, Muskelkrämpfe |
|
Erkrankungen der Nieren und Harnwege Häufig: Gelegentlich: |
seltenes Wasserlassen, Miktionsbeschwerden, Dysurie, Nykturie, Oligurie Spasmen der Harnblase, Hämaturie, erhöhte Harnfrequenz, Harnretention |
|
Erkrankungen der Geschlechtsorgane und der Brustdrüse Häufig: |
Druckempfindlichkeit der Brust, Hodenatrophie, |
|
Gelegentlich: |
Hodenschmerzen, Unfruchtbarkeit, Brusthypertrophie Gynäkomastie, Impotenz, Hodenerkrankung |
|
Selten: |
Schmerzen in der Brust |
|
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort Sehr häufig: |
Müdigkeit, Brennen an der Injektionsstelle, |
|
Häufig: |
Parästhesie an der Injektionsstelle Unwohlsein, Schmerzen an der Injektionsstelle, |
|
Gelegentlich: |
Bluterguss an der Injektionsstelle, Stechen an der Injektionsstelle, Rigor, Schwäche Juckreiz an der Injektionsstelle, Lethargie, |
|
Selten: |
Schmerzen, Fieber Ulzeration an der Injektionsstelle |
|
Sehr selten: |
Nekrose an der Injektionsstelle |
|
Erkrankungen des Blutes und des Lymphsystems Häufig: |
hämatologische Veränderungen |
|
Untersuchungen Häufig: |
Erhöhung der Kreatininphosphokinase im Blut, |
|
Gelegentlich: |
Verlängerung der Gerinnungszeit Erhöhung der Alaninaminotransferase, Erhöhung |
|
der Bluttriglyceride, Verlängerung der Prothrombinzeit, Gewichtszunahme |
Zu anderen unerwünschten Ereignissen, über die im Zusammenhang mit einer Leuprorelinacetat-Behandlung gewöhnlich berichtet wird, gehören periphere Ödeme, Lungenembolie, Palpitation, Myalgie, Muskelschwäche, veränderte Hautsensibilität, Schüttelfrost, peripherer Schwindel, Hautausschlag, Amnesie und Sehstörungen. In seltenen Fällen wurde nach Verabreichung von GnRH-Agonisten mit Kurz- oder Langzeitwirkung über einen Infarkt einer bereits bestehenden Hypophysenapoplexie berichtet. Über Thrombozytopenie und Leukopenie wurde in seltenen Fällen berichtet. Über Veränderungen der Glucosetoleranz liegen Berichte vor.
Die nach Injektion von ELIGARD berichteten unerwünschten Ereignisse am Verabreichungsort entsprechen den im Zusammenhang mit ähnlichen subkutan injizierten Präparaten beschriebenen unerwünschten Ereignissen.
Im Allgemeinen werden diese lokal begrenzten unerwünschten Ereignisse nach s. c. Injektionen als leicht und kurzzeitig beschrieben.
Veränderungen der Knochendichte
Bei Männern wurde in der Fachliteratur über eine Verminderung der Knochendichte infolge einer Orchiektomie oder einer Behandlung mit GnRH-Analoga berichtet. Daher ist unter einer Langzeittherapie mit Leuprorelin mit einer Verstärkung von Osteoporosesymptomen zu rechnen. Bezüglich des erhöhten Frakturrisikos infolge einer Osteoporose siehe Abschnitt 4.4.
Verschlechterung der Zeichen und Symptome der Erkrankung
In den ersten Wochen einer Behandlung mit Leuprorelinacetat kann es zu einer Verschlechterung der Zeichen und Symptome der Erkrankung kommen. Wenn sich Erkrankungen wie Wirbelsäulenmetastasen und/oder Harnwegsobstruktion oder Hämaturie verschlechtern, können neurologische Probleme wie Schwäche und/oder Parästhesie der unteren Extremitäten oder Verschlechterung der Harnwegsymptome auftreten.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Arzneimittel und Medizinprodukte, Abt. Pharmakovigilanz, Kurt-Georg-Kiesinger-Allee 3, D-53175 Bonn, Website: http://www.bfarm.de anzuzeigen.
4.9 Überdosierung
ELIGARD 7,5 mg hat kein Missbrauchspotenzial, und eine absichtliche Überdosierung ist unwahrscheinlich. Es gibt keine Berichte über Missbrauch oder Überdosierung unter Leuprorelinacetat in der klinischen Praxis. Sollte es dennoch zu einer Überdosierung mit Leuprorelinacetat kommen, sind die Beobachtung des Patienten und eine supportive symptomatische Therapie zu empfehlen.
5. PHARMAKOLOGISCHE EIGENSCHAFTEN
5.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Gonadotropin-Releasing-Hormon-Analoga, ATC-Code: L02A E02
Leuprorelinacetat ist ein synthetischer Nonapeptidagonist des natürlich vorkommenden Gonadotropin-Releasing-Hormons (GnRH), das bei kontinuierlicher Gabe die Gonadotropin-Sekretion der Hypophyse hemmt und beim Mann die testikuläre Steroidbildung supprimiert. Diese Wirkung ist nach Beendigung der Arzneitherapie reversibel. Allerdings übt der Agonist eine stärkere Wirkung aus als das natürliche Hormon, und die Zeitspanne bis zur Erholung der Testosteronspiegel kann von Fall zu Fall verschieden sein.
Die Verabreichung von Leuprorelinacetat verursacht zunächst einen Anstieg der Konzentrationen des luteinisierenden Hormons (LH) und des follikelstimulierenden Hormons (FSH) im Blutkreislauf, was beim Mann zu einer vorübergehenden Erhöhung der Blutspiegel der in den Gonaden produzierten Steroide (Testosteron und Dihydrotestosteron) führt. Bei kontinuierlicher Gabe von Leuprorelinacetat kommt es zu einem Absinken der LH- und FSH-Konzentrationen. Beim Mann sinkt der Testosteronspiegel innerhalb von 3 bis 5 Wochen nach Behandlungsbeginn unter die Kastrationsschwelle ab (< 50 ng/dl). Nach 6 Monaten liegt der mittlere Testosteronspiegel bei 6,1 (± 0,4) ng/dl und damit in einem Bereich wie nach bilateraler Orchiektomie. In der pivotalen klinischen Studie erreichten alle Patienten nach 6 Wochen den Kastrationsbereich (94 % am 28. und 98 % am 35. Tag). In den meisten Fällen lagen die Testosteronspiegel unter 20 ng/dl. Über den vollen Nutzen dieser niedrigen Konzentrationen liegen aber noch keine gesicherten Erkenntnisse vor. Die PSA-Spiegel sanken innerhalb von 6 Monaten um 94 % ab.
Langzeitstudien haben gezeigt, dass die Testosteronkonzentrationen bei kontinuierlicher Therapie bis zu 7 Jahre lang (wahrscheinlich zeitlich unbegrenzt) unterhalb des Kastrationsspiegels bleiben.
Die Tumorgröße wurde während dieser klinischen Versuche nicht direkt gemessen. Allerdings war mit einem Absinken des mittleren PSA-Spiegels um 94 % unter ELIGARD 7,5 mg indirekt eine positive Wirkung auf den Tumor zu erkennen.
In einer randomisierten klinischen Phase-III-Studie mit 970 Patienten mit lokal fortgeschrittenem Prostatakarzinom (hauptsächlich T2c bis T4, einige T1c bis T2b Patienten mit regionären Lymphknotenmetastasen) erhielten 483 Patienten eine KurzzeitAndrogenentzugstherapie (6 Monate) in Kombination mit Radiotherapie und 487 Patienten eine Langzeittherapie (3 Jahre). In einer Analyse auf Nichtunterlegenheit wurde die begleitende und adjuvante Kurzzeithormontherapie mit GnRH Agonisten (Triptorelin und Goserelin) mit der Langzeithormontherapie verglichen. Die Gesamtsterblichkeit nach 5 Jahren betrug 19,0 % in der Gruppe, die die Kurzzeittherapie erhielt, bzw. 15,2 % in der Gruppe, die die Langzeittherapie erhielt. Die beobachtete Hazard-Ratio von 1,42 mit einem oberen einseitigen 95,71 %-KI von 1,79 oder einem zweiseitigen 95,71 %-KI von 1,09; 1,85 (p = 0,65 für Nichtunterlegenheit), zeigt, dass die Kombination von Radiotherapie mit Androgenentzugstherapie über 6 Monate bezüglich des Überlebens gegenüber der Radiotherapie in Kombination mit Androgenentzugstherapie über 3 Jahre unterlegen ist. Das Gesamtüberleben nach 5 Jahren bei Langzeitbehandlung zeigte eine Überlebensrate von 84,8 % bzw. bei Kurzzeitbehandlung von 81,0 %. Die Lebensqualität, erfasst nach QLQ-C30, zeigte keine signifikanten Unterschiede zwischen den beiden Gruppen (P = 0,37). Die Ergebnisse gehen überwiegend auf die Patientenpopulation mit lokal fortgeschrittenen Tumoren zurück.
Der Nachweis für den Einsatz bei lokalisiertem Hochrisiko-Prostatakarzinom beruht auf veröffentlichten Studien zur Radiotherapie in Kombination mit GnRH-Analoga einschließlich Leuprorelinacetat. Es wurden klinische Daten aus fünf veröffentlichten Studien analysiert (EORTC 22863, RTOG 85-13, RTOG 92-02, RTOG 8610 und D’Amico et al., JAMA, 2004), die alle den Vorteil der Kombination von GnRH-Analoga mit Radiotherapie zeigen. Eine klare Differenzierung zwischen den entsprechenden Studienpopulationen für die Indikationen lokal fortgeschrittenes Prostatakarzinom und lokales Hochrisiko-Prostatakarzinom war in den veröffentlichten Studien nicht möglich.
Klinische Daten haben gezeigt, dass Radiotherapie gefolgt von 3 Jahren Androgenentzugstherapie gegenüber Radiotherapie gefolgt von 6 Monaten Androgenentzugstherapie vorzuziehen ist.
Die in medizinischen Leitlinien empfohlene Dauer der Androgenentzugstherapie für T3 bis T4 Patienten, die eine Radiotherapie erhalten, beträgt 2 bis 3 Jahre.
5.2 Pharmakokinetische Eigenschaften
Resorption: Bei Patienten mit fortgeschrittenem Prostatakarzinom steigen die mittleren Leuprorelinkonzentrationen im Serum nach einer initialen Injektion innerhalb von 4 bis 8 Stunden auf 25,3 ng/ml an (Cmax). Nach dem anfänglichen Anstieg (Plateauphase 2 bis 28 Tage nach jeder Gabe) bleiben die Serumkonzentrationen relativ konstant (0,28 - 1,67 ng/ml). Anzeichen für eine Akkumulation nach wiederholter Gabe sind nicht zu erkennen.
Verteilung: Bei gesunden männlichen Probanden beträgt das mittlere Steady-State-Volumen der Verteilung von Leuprorelin nach i. v. Bolusinjektion 27 l. In vitro lag die Bindung an humane Plasmaproteine zwischen 43 und 49 %.
Ausscheidung: Bei gesunden männlichen Probanden ergab die i.v. Bolusinjektion von 1 mg Leuprorelinacetat eine mittlere systemische Clearance von 8,34 l/h, mit einer terminalen Halbwertzeit der Ausscheidung von ca. 3 Stunden auf der Grundlage eines Zwei-Kompartiment-Modells.
Es wurden keine Studien zur Ausscheidung von ELIGARD durchgeführt.
Es wurden keine Studien zum Metabolismus von ELIGARD durchgeführt.
5.3 Präklinische Daten zur Sicherheit
In präklinischen Studien mit Leuprorelinacetat waren bei beiden Geschlechtern Wirkungen auf das Fortpflanzungssystem zu beobachten, die aufgrund der bekannten pharmakologischen Wirkungen dieser Substanz zu erwarten waren. Diese Wirkungen erwiesen sich nach Beendigung der Behandlung und nach einer entsprechenden Regenerationsphase als reversibel. Leuprorelinacetat zeigte keine teratogene Wirkung. Entsprechend den pharmakologischen Wirkungen von Leuprorelinacetat auf das Fortpflanzungssystem war beim Kaninchen eine Embryotoxizität/Letalität zu verzeichnen.
Kanzerogenitätsstudien wurden über einen Zeitraum von 24 Monaten an der Ratte und der Maus durchgeführt. Bei der Ratte war nach der s. c. Behandlung mit 0,6 - 4 mg/kg/d eine dosisabhängige Zunahme der Hypophysenapoplexie zu beobachten. Bei der Maus war hingegen kein solcher Effekt zu erkennen.
In einer Reihe von In-vitro- und In-vivo-Versuchen entfalteten Leuprorelinacetat und das Präparat ELIGARD 7,5 mg (einmonatige Wirkungsdauer) keine mutagene Wirkung.
6. PHARMAZEUTISCHE ANGABEN
6.1 Liste der sonstigen Bestandteile
Lösungsmittel (Spritze A): Poly(glycolsäure-co-milchsäure) (50 : 50)
N-Methylpyrrolidon (Ph.Eur.)
Pulver (Spritze B): keine
6.2 Inkompatibilitäten
Das in Spritze B vorliegende Leuprorelinacetat darf, außer mit dem Lösungsmittel in Spritze A, nicht mit anderen Arzneimitteln gemischt werden.
6.3 Dauer der Haltbarkeit
2 Jahre
Nach erstmaliger Entnahme dieses Produktes aus dem Kühlschrank kann es in der Originalverpackung bei Raumtemperatur (unter 25 °C) bis zu vier Wochen gelagert werden.
Nach dem erstmaligen Öffnen der Schalenverpackung sind Pulver und Lösungsmittel zur Herstellung einer Injektionslösung sofort zu mischen und zu verabreichen.
Nach dem Mischen sofort verwenden, da die Viskosität der Lösung mit der Zeit zunimmt.
6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Im Kühlschrank lagern (2 °C - 8 °C). In der Originalverpackung aufbewahren, um den Inhalt vor Feuchtigkeit zu schützen.
Dieses Produkt muss vor der Injektion auf Raumtemperatur gebracht werden. Ungefähr 30 Minuten vor der Anwendung aus dem Kühlschrank nehmen. Nach erstmaliger Entnahme aus dem Kühlschrank kann dieses Produkt in der Originalverpackung bei Raumtemperatur (unter 25 °C) bis zu vier Wochen gelagert werden.
6.5 Art und Inhalt des Behältnisses
Zwei vorgefüllte Cyclo-Olefin-Copolymer-/Polypropylen-Spritzen: Spritze B enthält das Pulver, Spritze A das Lösungsmittel. Die beiden Spritzen bilden zusammen ein Mischsystem.
Spritze A hat eine Kolbenspitze aus thermoplastischem Kautschuk und eine Luer-Lok-Schutzkappe aus Polyethylen oder Polypropylen. Die Verschlusskappe und die beiden Kolbenspitzen der Spritze B sind aus Chlorbutylkautschuk.
Folgende Packungsgrößen sind erhältlich:
• Packungen mit 2 thermoplastisch geformten Schalen in einem Umkarton. Eine der Schalen enthält eine vorgefüllte Polypropylenspritze (A), einen langen Kolben und einen Beutel mit Trockenmittel, die andere eine vorgefüllte Cyclo-Olefin-Copolymer-Spritze (B), eine sterile 20G-Nadel und einen Beutel mit Trockenmittel.
• Bündelpackung mit 3 x 2 vorgefüllten Polypropylen-/Cyclo-Olefin-Copolymer-Spritzen (eine Spritze A und eine Spritze B).
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung
Das Produkt auf Raumtemperatur anwärmen lassen indem Sie es ungefähr 30 Minuten vor der Anwendung aus dem Kühlschrank nehmen. Zuerst ist der Patient für die Injektion vorzubereiten. Danach ist die Injektionslösung wie unten beschrieben herzustellen. Falls bei der Zubereitung des Produktes nicht korrekt vorgegangen wurde, darf es nicht verabreicht werden, da mangelnde klinische Wirksamkeit aufgrund fehlerhafter Rekonstitution des Produktes auftreten kann.
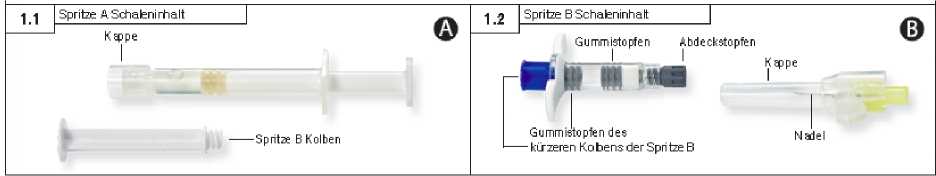
1. Schritt: Beide Schalenverpackungen durch Abreißen der Folie an der durch eine kleine Blase gekennzeichneten Ecke öffnen und den Inhalt (2 Schalen mit Spritze A [Abb. 1.1] bzw. Spritze B [Abb. 1.2]) auf einer sauberen Arbeitsfläche auslegen. Beutel mit dem Trockenmittel entsorgen.
2. Schritt: Blau gefärbten, kurzen Kolben zusammen mit angehängtem grauen Stopfen von Spritze B abziehen (nicht abschrauben) und entsorgen (Abb. 2). Das Produkt kann nicht gemischt werden, wenn sich 2 graue Stopfen in Spritze B befinden.

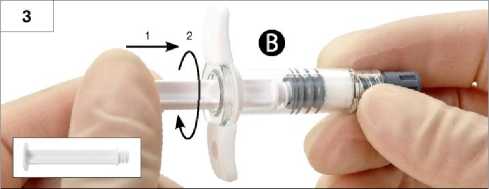
3. Schritt: Weißes Kolbenstück der Spritze B vorsichtig auf den verbliebenen grauen Stopfen in Spritze B schrauben (Abb. 3).
4. Schritt: Graue Gummikappe von Spritze B entfernen und Spritze ablegen (Abb. 4).

5. Schritt: Spritze A senkrecht halten, um ein Auslaufen der Flüssigkeit zu vermeiden, und die helle Verschlusskappe dieser Spritze abschrauben (Abb. 5).

6. Schritt: Die beiden Spritzen ineinanderschieben und Spritze B auf Spritze A bis zum Anschlag aufschrauben (Abb. 6 a und b). Nicht überdrehen.

7. Schritt: Die zusammengeschraubte Einheit umdrehen und die Spritzen weiterhin senkrecht halten (Spritze B unten) und die in der Spritze A enthaltene Flüssigkeit in die Spritze B mit dem darin enthaltenen Pulver (Leuprorelinacetat) hineindrücken (Abb. 7).

8. Schritt: Mischen Sie das Produkt sorgfältig, indem Sie die Spritzen waagerecht halten und den Spritzeninhalt beider Spritzen sanft zwischen den Spritzen hin- und her-bewegen (insgesamt ca. 60-mal, dauert ungefähr 60 Sekunden). So erhalten Sie eine homogene, viskose Lösung (Abb. 8). Die zusammengeschraubten Spritzen nicht verbiegen, da die Spritzen dadurch eventuell leicht auseinandergeschraubt werden, was zum Auslaufen von Flüssigkeit führen kann.

Nach sorgfältigem Vermischen entsteht eine viskose, farblose bis weiße oder blassgelbe Lösung (weiße bis blassgelbe Farbtöne sind möglich).
Wichtig: Nach Mischen sofort mit nächstem Schritt fortfahren, da die Viskosität der Lösung, wenn sie einmal hergestellt ist, mit der Zeit zunimmt. Hergestelltes Produkt nicht weiter kühlen.
Anmerkung: Das Präparat muss nach diesem Verfahren gemischt werden. Durch Schütteln lässt sich KEINE verwendbare Mischung herstellen.
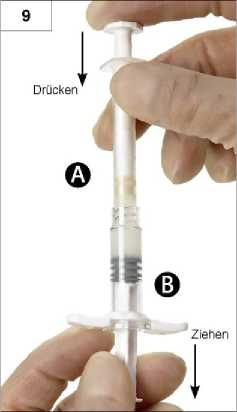
9. Schritt: Spritzen senkrecht halten (Spritze B unten). Die Spritzen müssen fest aneinandergeschraubt sein. Gesamtes Gemisch durch Druck auf den Kolben der Spritze A und leichtes Zurückziehen des Kolbens der Spritze B in die kurze, breitere Spritze B drücken (Abb. 9).
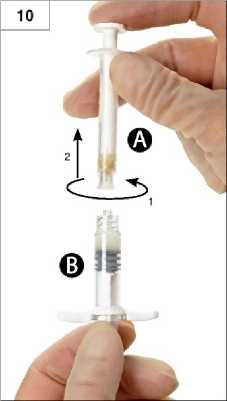
10. Schritt: Spritze A abschrauben, während der Kolben der Spritze A weiterhin nach unten gedrückt wird (Abb. 10). Es darf keine Flüssigkeit auslaufen, weil sonst die Nadel nicht sicher schließend aufgeschraubt werden kann.
Anmerkung: Eine große oder mehrere kleine Luftblasen können im Präparat verbleiben und stellen kein Problem dar.
Bitte in dieser Phase keine Luftblasen aus Spritze B ausdrücken, da es zu einem Produktverlust kommen kann.
11. Schritt: Spritze B senkrecht halten. Öffnen Sie die Verpackung der Sicherheitsnadel durch Aufreißen an der Lasche der Papierfolie und entnehmen Sie die Sicherheitsnadel. Anschließend die Sicherheitsnadel an Spritze B durch Halten der Nadel und Drehen der Spritze im Uhrzeigersinn anbringen, damit die Nadel vollständig an der Spritze sitzt (Abb. 11). Nicht überdrehen.

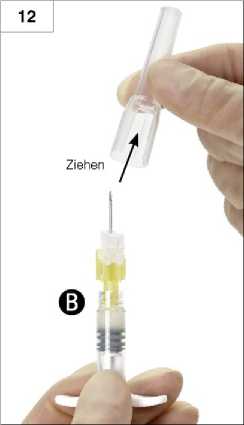
12. Schritt: Vor Injektion Schutzkappe der Nadel entfernen (Abb. 12).
Wichtig: Vor Injektion sicherstellen, dass die Schutzvorrichtung der Sicherheitsnadel nicht betätigt wird.
13. Schritt: Vor der Anwendung große Luftblasen aus der Spritze B entfernen. Applizieren Sie das Produkt subkutan. Stellen Sie sicher, dass der komplette Inhalt aus Spritze B injiziert wird.
14. Schritt: Schließen Sie die Schutzvorrichtung nach der Applikation auf eine der folgenden Weisen. 1. Verschließen auf flacher Oberfläche
Drücken Sie die Schutzvorrichtung mit dem Hebel nach unten auf eine glatte Oberfläche (Abb. 14.1 a und b), um die Nadel zu bedecken, und schließen Sie die Schutzvorrichtung.
Ein hör- und fühlbares „Klick“ zeigt an, dass die Schutzvorrichtung richtig geschlossen wurde. Eine geschlossene Schutzvorrichtung umschließt die Nadel vollständig (Abb. 14.1 b).

2. Verschließen mit Daumen
Schieben Sie mit dem Daumen die Schutzvorrichtung Richtung Nadelspitze (Abb. 14.2 a und b), um die Nadel zu bedecken, und schließen Sie die Schutzvorrichtung.
Ein hör- und fühlbares „Klick“ zeigt an, dass die Schutzvorrichtung richtig geschlossen wurde. Eine geschlossene Schutzvorrichtung umschließt die Nadel vollständig (Abb. 14.2 b).

15. Schritt: Sobald die Schutzvorrichtung verschlossen ist, sofort Nadel und Spritze in einem passenden Spritzenbehälter entsorgen.
7. INHABER DER ZULASSUNG
Astellas Pharma Europe B.V. Sylviusweg 62 2333 BE Leiden Niederlande
Mitvertreiber
Astellas Pharma GmbH Postfach 50 01 66 80971 München Tel.: (089) 45 44 01 Fax: (089) 45 44 13 29 EMail: info.de@astellas.com
Falls weitere Informationen über das Arzneimittel gewünscht werden, setzen Sie sich bitte mit dem Mitvertreiber in Verbindung.
8. ZULASSUNGSNUMMER(N)
53653.00.00
9. DATUM DER ERTEILUNG DER ZULASSUNG/VERLÄNGERUNG DER ZULASSUNG
Datum der Erteilung der Zulassung: 26. November 2003 Datum der letzten Verlängerung der Zulassung: 03. Mai 2011
10. STAND DER INFORMATION
04/2015
11. VERKAUFSABGRENZUNG
Verschreibungspflichtig