Feiba Nf 500 E
FEIBA Fachinformation
1. BEZEICHNUNG DES ARZNEIMITTELS
FEIBA NF 500 E/1000 E
Pulver und Lösungsmittel zur Herstellung einer Injektionslösung.
2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
Wirkstoff: Eine mit Faktor VIII-Inhibitor-Bypassing-Aktivität angereicherte Humanplasmafraktion 1 Durchstechflasche Pulver enthält
|
Arzneimittel |
FEIBA NF 500 E |
FEIBA NF 1000 E |
|
Humanplasmaprotein |
200 - 600 mg |
400 - 1200 mg |
|
mit einer | ||
|
Faktor VIII-Inhibitor-Bypassing-Aktivität von |
500 E* |
1000 E* |
|
Aktivität pro ml |
25 E |
50 E |
* 1 FEIBA-Einheit (Factor Eight Inhibitor Bypassing Activity) verkürzt die aktivierte partielle Thromboplastinzeit eines hochtitrigen Faktor VIII Inhibitorplasmas (Hausstandard) auf 50 % des Puffer-Leerwertes.
FEIBA enthält Faktor II, IX und X in vorwiegend nicht aktivierter Form sowie aktivierten Faktor VII; das Antigen Gerinnungsfaktor VIII (F VIII C:Ag) ist in einer Konzentration von bis zu 0,1 E/1 E FEIBA vorhanden. Die Faktoren des Kallikrein-Kinin-Systems sind nur in Spuren vorhanden.
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
3. DARREICHUNGSFORM
Pulver und Lösungsmittel zur Herstellung einer Injektionslösung.
Weiße, fast weiße oder hellgrüne gefriergetrocknete pulvrige oder kompakte Trockensubstanz.
Der pH-Wert der rekonstituierten Lösung liegt zwischen 6,8 und 7,6. Die Osmolalität der rekonstituierten Lösung ist >240 mOsmol/kg.
4. KLINISCHE ANGABEN
4.1 Anwendungsgebiete
• Behandlung und Prophylaxe von Blutungen bei Hämophilie-A-Patienten mit FVIII-Inhibitor
• Behandlung und Prophylaxe von Blutungen bei Hämophilie-B-Patienten mit FIX-Inhibitor
• Behandlung und Prophylaxe von Blutungen bei nicht Hämophiliekranken mit einem erworbenen Inhibitor gegen die Faktoren VIII, IX oder XI.
In einzelnen Fällen wurde FEIBA erfolgreich bei von-Willebrand-Patienten mit einem Inhibitor eingesetzt.
FEIBA wurde außerdem in Kombination mit Faktor VIII-Konzentrat für eine Langzeittherapie eingesetzt, um eine vollständige und dauerhafte Eliminierung des FVIII-Inhibitors zu erreichen und so eine regelmäßige Behandlung mit FVIII-Konzentrat wie bei Patienten ohne Inhibitor zu ermöglichen.
4.2 Dosierung und Art der Anwendung
Dosierung
Die Behandlung sollte unter der Aufsicht eines mit der Therapie der Hämophilie erfahrenen Arztes eingeleitet werden.
Die Dosierung und Dauer der Therapie hängen von der Schwere der Hämostasestörung sowie dem Ort und dem Ausmaß der Blutung und dem klinischen Zustand des Patienten ab.
Dosierung und Dosierungsintervall sollten sich immer nach der klinischen Wirksamkeit im Einzelfall richten.
Als allgemeine Richtlinie wird eine Dosis von 50 bis 100 E FEIBA pro kg Körpergewicht empfohlen; es sollte jedoch eine Einzeldosis von 100 E/kg Körpergewicht und eine Tagesdosis von 200 E/kg Körpergewicht nicht überschritten werden, es sei denn, die Anwendung von höheren Dosen ist durch die Schwere der Blutung erforderlich und gerechtfertigt. Siehe Abschnitt 4.4.
Kinder und Jugendliche
Die Erfahrung bei Kindern unter 6 Jahren ist begrenzt; die Dosierung sollte sich an den Empfehlungen für Erwachsene orientieren und ist an den klinischen Zustand des jeweiligen Kindes anzupassen.
1) Spontane Blutung
Gelenks-, Muskel- und Weichteilblutungen
Für leichte bis mittelschwere Blutungen wird eine Dosis von 50 - 75 E/kg Körpergewicht in zwölfstündigen Intervallen empfohlen. Die Behandlung sollte bis zum Eintreten einer eindeutigen Besserung der klinischen Symptome, wie Nachlassen der Schmerzen, Abnahme der Schwellung oder Mobilisierung des Gelenks, erfolgen.
Für schwere Blutungen und Weichteilblutungen, wie zum Beispiel retroperitoneale Blutungen, wird eine Dosis von 100 E/kg Körpergewicht in zwölfstündigen Intervallen empfohlen.
Schleimhautblutungen
Die Verabreichung einer Dosis von 50 E/kg Körpergewicht alle 6 Stunden bei sorgfältiger Überwachung des Patienten wird empfohlen (visuelle Überwachung der Blutung, regelmäßige Hämatokrit-Messungen). Wird die Blutung nicht gestillt, kann die Dosis auf 100 E/kg Körpergewicht erhöht werden. Dabei darfjedoch die maximale Tagesdosis von 200 E/kg Körpergewicht nicht überschritten werden.
Andere schwere Blutungen
Schwere Blutungen, wie ZNS-Blutungen, wurden erfolgreich mit Dosen von 100 E/kg Körpergewicht in zwölfstündigen Intervallen behandelt. In Einzelfällen kann eine Verkürzung der Applikationsintervalle auf 6 Stunden erforderlich sein, bis eine eindeutige klinische Besserung erreicht wird. Dabei darf jedoch die maximale Tagesdosis von 200 E/kg Körpergewicht nicht überschritten werden.
2) Chirurgische Eingriffe
50 - 100 E/kg Körpergewicht sollten - unter Beachtung der maximalen Tagesdosis - in Intervallen von bis zu 6 Stunden verabreicht werden.
3) Prophylaxe
• Blutungsprophylaxe bei Patienten mit hohen Inhibitorwerten und häufigen Blutungen, bei denen eine Immuntoleranztherapie (ITT) gescheitert ist oder nicht in Betracht kommt.
Es wird eine Dosis von 50 - 100 E/kg Körpergewicht dreimal wöchentlich empfohlen. Diese Dosis darf bis auf 100 E/kg Körpergewicht jeden Tag gesteigert werden, wenn der Patient weiterhin blutet oder darf allmählich gesenkt werden.
• Blutungsprophylaxe bei Patienten mit hohen Inhibitorwerten unter ITT (Immuntoleranztherapie): FEIBA kann in Kombination mit FVIII-Konzentrat in Dosierungen von 50 - 100 E/kg Körpergewicht ein- bis zweimal täglich verabreicht werden, bis der Faktor VIII-Inhibitor auf <2 B.E. reduziert wurde.
Überwachung
Wegen des komplexen Wirkmechanismus ist eine unmittelbare Überwachung der Wirkstoffe nicht möglich. Gerinnungstests, wie zum Beispiel die Vollblutgerinnungszeit, das Thromboelastogramm (TEG, r-Wert) und die aPTT weisen gewöhnlich nur eine geringe Verkürzung auf und spiegeln nicht unbedingt die klinische Wirksamkeit wider. Daher haben diese Tests beim Monitoring der FEIBA-Therapie nur eine geringe Bedeutung. Bei ungenügendem Ansprechen auf die Therapie wird empfohlen, die Thrombozytenzahl zu bestimmen. Siehe Abschnitt 4.4.
Art der Anwendung
FEIBA wird als intravenöse Injektion oder Infusion verabreicht. Die Injektions-/Infusionsrate ist so zu wählen, dass sie für den Patienten möglichst angenehm ist und 2 Einheiten FEIBA pro kg Körpergewicht in der Minute nicht überschritten werden. Hinweise zur Rekonstitution des Arzneimittels vor der Anwendung, siehe Abschnitt 6.6.
4.3 Gegenanzeigen
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
FEIBA darf in den folgenden Situationen nicht angewendet werden, wenn therapeutische Alternativen zu FEIBA zur Verfügung stehen:
• Disseminierte intravasale Koagulation (DIC, Verbrauchskoagulopathie)
• Akute Thrombose oder Embolie (einschließlich Myokardinfarkt)
Siehe Abschnitt 4.4.
1 Bethesda-Einheit ist definiert als die Menge an Antikörpern, die 50 % der FVIII-Aktivität von frischem durchschnittlichem Humanplasma nach einer zweistündigen Inkubation bei 37 °C hemmt.
4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Warnhinweise
Thrombosen und thromboembolische Ereignisse
Thrombosen und thromboembolische Ereignisse (TTE), z. B. disseminierte intravasale Koagulation (DIC), Venenthrombose, Lungenembolie, Myokardinfarkt und Schlaganfall, sind im Verlauf der Behandlung mit FEIBA aufgetreten.
Die begleitende Behandlung mit rekombinantem Faktor VIIa erhöht wahrscheinlich das Risiko für das Auftreten von TTE. Das Risiko von TTE kann auch bei hohen FEIBA-Dosen erhöht sein. Einige dieser Ereignisse traten bei täglichen Dosen über 200 E/kg Körpergewicht, bei Patienten mit anderen Risikofaktoren für TTE (einschließlich DIC, fortgeschrittener Atherosklerose, Quetschtrauma oder Septikämie) auf. Bei Patienten mit angeborener oder erworbener Hämophilie sollte stets in Betracht gezogen werden, dass möglicherweise solche Risikofaktoren bestehen. Siehe Abschnitt „Überwachung der Therapie“.
Bei Patienten, bei denen ein Risiko für eine DIC oder eine arterielle oder venöse Thrombose besteht, sollte FEIBA mit besonderer Vorsicht angewendet werden. Siehe Abschnitt 4.3.
Bei den ersten Anzeichen von TTE ist die Infusion unverzüglich abzubrechen und es sind geeignete diagnostische und therapeutische Maßnahmen einzuleiten.
Allergische Überempfindlichkeitsreaktionen
FEIBA kann allergische Überempfindlichkeitsreaktionen, einschließlich Urtikaria, Angioödem, gastrointestinale Manifestationen, Bronchospasmen und Hypotonie auslösen; diese Reaktionen können einen schweren und systemischen Verlauf nehmen (z. B. Anaphylaxie mit Urtikaria und Angioödem, Bronchospasmen und Kreislaufschock). Auch andere Infusionsreaktionen wie Schüttelfrost, Fieber und Hypertonie wurden berichtet.
Beim ersten Anzeichen oder Symptom einer Infusions- oder Überempfindlichkeitsreaktion ist die Anwendung von FEIBA unverzüglich abzubrechen und es ist eine geeignete medizinische Versorgung einzuleiten.
Wenn bei Patienten mit bekannter Überempfindlichkeit oder Verdacht auf Überempfindlichkeit gegen das Produkt oder einen der Inhaltsstoffe eine Re-Exposition gegenüber FEIBA in Betracht gezogen wird, ist eine sorgfältige Abwägung des erwarteten Nutzens einer Re-Exposition gegenüber den Risiken erforderlich. Dabei sind sowohl die Art der bekannten oder vermuteten Überempfindlichkeit des Patienten (allergisch oder nicht-allergisch), die potenziellen Maßnahmen zur Behandlung und/oder Prävention, sowie alternative Therapeutika zu berücksichtigen.
Überwachung der Therapie
Eine Einzeldosis von 100 E/kg Körpergewicht und eine Tagesdosis von 200 E/kg Körpergewicht sollten nicht überschritten werden. Patienten, die eine Dosis von mehr als 100 E/kg Körpergewicht erhalten, müssen auf Anzeichen einer sich entwickelnden Thrombose oder eines thromboembolischen Ereignisses, z.B. DIC oder akute Koronarischämie hin überwacht werden. Hohe Dosierungen von FEIBA sollten nur bei absoluter Notwendigkeit zum Stillen von Blutungen verabreicht werden.
Im Fall von klinisch relevanten Veränderungen des Blutdrucks und der Pulsrate, bei Atembeschwerden, Schmerzen in der Brust sowie Husten, sollte die Infusion sofort unterbrochen und entsprechende
diagnostische und therapeutische Maßnahmen eingeleitet werden. Niedrige Fibrinogenwerte, ein gesenkter Thrombozytenspiegel und/oder das Vorhandensein von Fibrin/Fibrinogen-Abbauprodukten sind Laborergebnisse, die auf DIC hindeuten. Weitere Hinweise auf DIC sind unter anderem eine wesentlich verlängerte Thrombinzeit, Prothrombinzeit oder aPTT.
Nicht hämophiliekranke Patienten
Nicht hämophiliekranke Patienten mit einem erworbenen Inhibitor gegen Faktor VIII, IX oder XI können gleichzeitig eine erhöhte Anfälligkeit für Blutungen und ein erhöhtes Thromboserisiko aufweisen.
Labortests und klinische Wirksamkeit
In-vitro-Tests, wie zum Beispiel aPTT, Vollblutgerinnungszeit und das Thromboelastogramm (TEG) stimmen möglicherweise nicht mit dem klinischen Bild überein. Aus diesem Grund wird von Versuchen, diese Werte durch eine Erhöhung der FEIBA-Dosen zu normalisieren, entschieden abgeraten, da diese nicht nur erfolglos bleiben könnten, sondern dabei auch ein potenzielles Risiko einer DIC durch Überdosierung besteht.
Bedeutung des Thrombozytenspiegels
Im Fall einer ungenügenden Reaktion auf die Behandlung mit FEIBA wird empfohlen, den Thrombozytenspiegel zu bestimmen, da eine ausreichende Anzahl an intakten Thrombozyten für die Wirksamkeit von FEIBA benötigt wird.
Maßnahmen zum Schutz vor einer Übertragung von infektiösen Erregern
Standardmaßnahmen zur Vorbeugung von Infektionen, die sich durch den Einsatz von Arzneimitteln ergeben, die aus menschlichem Blut oder Blutplasma hergestellt sind, schließen die Auswahl der Spender und das Screening der einzelnen Blutspenden und Plasmapools auf spezifische Infektionsmarker sowie den Einsatz effektiver Schritte zur Inaktivierung/Eliminierung von Viren im Herstellungsverfahren ein. Dennoch kann bei der Verabreichung von Arzneimitteln aus menschlichem Blut oder Blutplasma die Möglichkeit der Übertragung von Krankheitserregern nicht völlig ausgeschlossen werden. Dies gilt auch für bislang unbekannte oder neu aufgetretene Viren und andere Pathogene.
Die getroffenen Maßnahmen werden als wirksam erachtet für umhüllte Viren wie das Immunschwächevirus (HIV), das Hepatitis B-Virus (HBV) und Hepatitis C-Virus (HCV), und für das nicht-umhüllte Hepatitis A-Virus. Für nicht-umhüllte Viren wie Parvovirus B19 können diese Maßnahmen möglicherweise nur begrenzt wirksam sein. Parvovirus B19-Infektionen können schwerwiegende Folgen für Schwangere (Infektion des Fötus) und für Menschen mit Immunmangelkrankheiten oder gesteigerter Erythropoese (z. B. hämolytische Anämie) haben.
Es wird dringend empfohlen, bei jeder Verabreichung von FEIBA den Produktnamen und die Chargennummer zu notieren, um die Rückverfolgbarkeit der verwendeten Charge sicherzustellen.
Bei Patienten, die regelmäßig oder wiederholt aus menschlichem Blutplasma hergestellte Präparate einschließlich FEIBA erhalten, müssen geeignete Impfungen (Hepatitis A und B) in Betracht gezogen werden.
Vorsichtsmaßnahmen
Thrombosen und Thromboembolische Komplikationen
In den folgenden Fällen sollte FEIBA nur verabreicht werden, wenn - zum Beispiel aufgrund eines sehr hohen Inhibitortiters oder bei einer Blutung oder einem Blutungsrisiko mit Lebensgefahr (z. B. posttraumatisch oder postoperativ) - kein Ansprechen auf die Behandlung mit dem passenden Gerinnungsfaktor-Konzentrat erwartet werden kann.
Disseminierte intravasale Koagulation (DIC):
• Laborsymptome und/oder klinische Symptome, die eindeutig auf DIC hinweisen.
• Laborzeichen, histologische und/oder klinische Anzeichen einer Leberschädigung; wegen der verspäteten Clearance der aktivierten Gerinnungsfaktoren besteht für diese Patienten ein erhöhtes Risiko einer DIC.
Myokardinfarkt, akute Thrombose und/oder Embolie:
• Bei vermuteter oder nachgewiesener koronarer Herzkrankheit sowie bei Patienten mit akuter Thrombose und/oder Embolie ist die Anwendung von FEIBA nur bei lebensbedrohlichen Blutungen indiziert.
Uneinheitliches Ansprechen auf Bypassing-Präparate
Wegen patientenspezifischer Faktoren können die Reaktionen auf ein Bypassing-Präparat unterschiedlich ausfallen. Patienten, die in einer Blutungssituation keine ausreichende Reaktion auf ein bestimmtes Präparat zeigen, können eventuell auf ein anderes Präparat ansprechen. Spricht ein Patient nicht ausreichend auf ein Bypassing-Präparat an, sollte ein anderes Präparat in Betracht gezogen werden.
Anamnestische Reaktionen
Die Gabe von FEIBA an Hemmkörper-Patienten kann zu einem vorübergehenden anamnestischen Anstieg des Hemmkörper-Titers führen. Im Verlauf der fortgesetzten FEIBA-Gabe kann der Hemmkörper-Titer wieder sinken. Die klinischen und publizierten Daten lassen darauf schließen, dass die Wirksamkeit von FEIBA dadurch nicht beeinträchtigt wird.
Antikörper gegen Hepatitis-B-Oberflächenantigen und Interpretation von Testergebnissen Nach Verabreichung hoher Dosen von FEIBA kann es aufgrund eines passageren Anstiegs des Titers an passiv übertragenen Antikörpern gegen Hepatitis-B-Oberflächenantigen zu Fehlinterpretationen von positiven Ergebnissen in serologischen Tests kommen.
Prophylaktische Anwendung
Es liegen nur in begrenztem Umfang klinische Daten zur Anwendung von FEIBA für die Blutungsprophylaxe bei Hämophilie-Patienten vor.
Anwendung bei Kindern
Zur Anwendung bei Kindern unter 6 Jahren liegen nur begrenzte Daten vor. Die aufgeführten Warnhinweise und Vorsichtsmaßnahmen gelten für Erwachsene und Kinder gleichermaßen.
Wichtige Information über bestimmte sonstige Bestandteile von FEIBA
FEIBA enthält ca. 80 mg Natrium (kalkuliert) pro Durchstechflasche. Dies sollte bei Patienten, die auf eine natriumarme Ernährung achten müssen, berücksichtigt werden.
4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Es wurden keine Studien zur Erfassung von Wechselwirkungen durchgeführt.
Die Verabreichung von Antifibrinolytika in Kombination mit FEIBA ist nicht zu empfehlen, da es bei gleichzeitiger Anwendung zu Thrombosen und thromboembolischen Ereignissen kommen kann.
Falls die gemeinsame Gabe von Antifibrinolytika und FEIBA indiziert ist, sollten die Medikamente im Abstand von mindestens 6 Stunden verabreicht werden.
Bei gleichzeitiger Gabe von rFVIIa lassen die verfügbaren in-vitro- und klinischen Daten darauf schließen, dass es zu Arzneimittelwechselwirkungen kommen könnte (z. B. mit der Folge von Thrombosen oder thromboembolischen Ereignissen).
4.6 Fertilität, Schwangerschaft und Stillzeit
Zur Sicherheit von FEIBA bei Anwendung in der Schwangerschaft und Stillzeit liegen keine Daten vor. Der Arzt muss die potenziellen Risiken abwägen und darf FEIBA nur bei eindeutiger Indikationsstellung verordnen. Dabei ist zu beachten, dass während der Schwangerschaft und der postpartalen Periode ein erhöhtes Risiko für thromboembolische Ereignisse besteht und zahlreiche Schwangerschaftskomplikationen mit einem erhöhten DIC-Risiko verbunden sind.
Es wurden keine Reproduktionsstudien an Tieren mit FEIBA durchgeführt und die Auswirkungen von FEIBA auf die Fertilität sind nicht in kontrollierten klinischen Prüfungen bestimmt worden.
4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
FEIBA hat keine Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
4.8 Nebenwirkungen
Die in diesem Abschnitt aufgeführten Nebenwirkungen wurden im Rahmen der Post-MarketingÜberwachung und in 2 klinischen Studien berichtet, in denen FEIBA in der Behandlung von Blutungsepisoden bei pädiatrischen und erwachsenen Patienten mit Hämophilie A oder B und mit Hemmkörpern gegen die Faktoren VIII oder IX angewendet wurde. In einer dieser Studien wurden auch Patienten mit erworbener Hämophilie mit Hemmkörpern gegen Faktor VIII eingeschlossen (2 von 49 Patienten).
Außerdem wurden Nebenwirkungen aus einer dritten Studie, in der die Prophylaxe mit einer Bedarfsbehandlung verglichen wurde, aufgenommen.
Die Häufigkeit der Nebenwirkungen wurde nach den folgenden Kriterien angegeben:
sehr häufig: häufig: gelegentlich: selten: sehr selten:
> 1/10
> 1/100 bis <1/10
> 1/1000 bis <1/100
> 1/10 000 bis <1/1000 < 1/10 000
nicht bekannt: Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar
|
Häufigkeit von Nebenwirkungen aus spontanen Meldungen und klinischen Studien | ||
|
Systemorganklassen gemäß MedDRA |
Nebenwirkung |
Häufigkeit* |
|
Erkrankungen des Blutes |
Disseminierte intravasale Gerinnung (DIC) |
nicht bekannt |
|
und des Lymphsystems |
Anstieg des Hemmkörper-Titers (anamnestische Reaktion) ,a | |
|
Erkrankungen des |
Überempfindlichkeitc |
häufig |
|
Immunsystems |
Nesselsucht |
nicht bekannt |
|
Anaphylaktische Reaktion |
nicht bekannt | |
|
Erkrankungen des |
Parästhesie |
nicht bekannt |
|
Nervensystems |
Hypästhesie (Taubheitsgefühl in den Gliedmaßen) |
nicht bekannt |
|
Schlaganfall thrombotischen Ursprungs |
nicht bekannt | |
|
Schlaganfall embolischen Ursprungs |
nicht bekannt | |
|
Kopfschmerzenc |
häufig | |
|
Somnolenz* |
nicht bekannt | |
|
Benommenheitb |
häufig | |
|
Geschmacksstörung* |
nicht bekannt | |
|
Herzerkrankungen |
Myokardinfarkt Tachykardie |
nicht bekannt |
|
Gefäßerkrankungen |
Thrombose |
nicht bekannt |
|
Venöse Thrombose |
nicht bekannt | |
|
Arterielle Thrombose |
nicht bekannt | |
|
Embolie (thromboembolische Komplikationen) |
nicht bekannt | |
|
Hypotoniec |
häufig | |
|
Hypertonie |
nicht bekannt | |
|
Hitzegefühl |
nicht bekannt | |
|
Erkrankungen der Atem- |
Lungenembolie | |
|
wege, des Brustraums |
Bronchospasmus | |
|
und Mediastinums |
Keuchende Atmung Husten Dyspnoe* |
nicht bekannt |
|
Erkrankungen des |
Erbrechen | |
|
Gastrointestinaltrakts |
Diarrhoe Abdominale Beschwerden Übelkeit* |
nicht bekannt |
|
Erkrankungen der Haut |
Taubheitsgefühl im Gesicht |
nicht bekannt |
|
und des Unterhaut- |
Angioödem |
nicht bekannt |
|
zellgewebes |
Nesselsucht |
nicht bekannt |
|
Juckreiz |
nicht bekannt | |
|
Hautausschlagc |
häufig | |
|
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort |
Schmerzen an der Injektionsstelle Unwohlsein Hitzegefühl Schüttelfrost* Pyrexie* Brustschmerzen* Unwohlsein im Brustbereich* |
nicht bekannt |
|
Untersuchungen |
Blutdruckabfall Positiver Test auf Antikörper gegen Hepatitis-B-Oberflächenantigenc |
nicht bekannt häufig |
*
Nebenwirkungen in Zusammenhang mit der Wirkstoffklasse
Andere Symptome einer Überempfindlichkeitsreaktion gegen Produkte, die aus Plasma hergestellt sind, sind Lethargie und Unruhe.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel, Paul-Ehrlich-Institut, Paul-Ehrlich-Str. 51-59, 63225 Langen, Tel: +49 6103 77 0, Fax: +49 6103 77 1234, Website: www.pei.de anzuzeigen.
4.9 Überdosierung
Das Risiko von Thrombosen und thromboembolischen Ereignissen (TTE, z.B. disseminierte intravasale Koagulation, Venenthrombose, Lungenembolie, Myokardinfarkt) kann bei hohen FEIBA-Dosen erhöht sein. Einige solcher Ereignisse traten bei täglichen Dosen über 200 E/kg Körpergewicht, bei Patienten mit anderen Risikofaktoren für TTE auf (siehe auch Abschnitt 4.4)
Bei ersten Anzeichen von TTE ist die Infusion unverzüglich abzubrechen und es sind geeignete diagnostische und therapeutische Maßnahmen einzuleiten (siehe Abschnitt 4.4).
5. PHARMAKOLOGISCHE EIGENSCHAFTEN
5.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: aktivierter Prothrombinkomplex gegen Faktor VIII-Antikörper, ATC-Code: B02BD03
Obwohl FEIBA in den frühen siebziger Jahren entwickelt wurde und seine Faktor VIII-Inhibitor-Bypassing-Aktivität sowohl in vitro als auch in vivo belegt wurde, ist sein Wirkprinzip immer noch Thema der wissenschaftlichen Diskussion. Neue wissenschaftliche Erkenntnisse deuten jedoch darauf hin, dass spezifische Komponenten des aktivierten Prothrombinkomplexes, das Proenzym Prothrombin (F II) und aktivierter Faktor X (F Xa), eine Rolle beim FEIBA-Wirkmodus spielen.
5.2 Pharmakokinetische Eigenschaften
Da das Wirkprinzip immer noch diskutiert wird, können keine eindeutigen Aussagen über die pharmakokinetischen Eigenschaften von FEIBA getroffen werden.
5.3 Präklinische Daten zur Sicherheit
Aus den Studien zur akuten Toxizität bei Faktor VIII-Knockout-Mäusen, normalen Mäusen und Ratten, mit Dosierungen, die die menschliche maximale Tagesdosis (d. h. >200 E/kg Körpergewicht) überschreiten, kann geschlossen werden, dass Nebenwirkungen von FEIBA vorwiegend die Folge von Hyperkoagulation durch die pharmakologischen Eigenschaften des Produkts sind.
Die in FEIBA enthaltenen humanen Plasmaproteine verhalten sich wie körpereigene Bestandteile. Die Untersuchungen an Labortieren mit einmaliger Dosierung ergaben keinen Hinweis auf ein pharmakologisch-toxikologisches Potential von FEIBA für die Anwendungen am Menschen. Toxizitätsstudien an Tieren mit mehrfacher Dosisgabe sind wegen immunologischer Reaktionen auf heterologe Proteine nicht sinnvoll durchzuführen.
Da Humanplasmaproteine offenbar keine kanzerogenen oder mutagenen Wirkungen aufweisen, werden experimentelle Studien, insbesondere bei heterologen Spezies, nicht als zwingend notwendig erachtet.
6. PHARMAZEUTISCHE ANGABEN
6.1 Liste der sonstigen Bestandteile
Pulver
Natriumchlorid
Natriumcitrat
Lösungsmittel
Wasser für Injektionszwecke
6.2 Inkompatibilitäten
Da keine Kompatibilitätsstudien durchgeführt wurden, darf FEIBA nicht mit anderen Arzneimitteln oder mit Lösungsmitteln gemischt werden. Es ist ratsam, einen gemeinsamen Venenzugang vor und nach der Infusion von FEIBA mit isotonischer Kochsalzlösung zu spülen.
Gerinnungsfaktoren aus Humanplasma können von den inneren Oberflächen bestimmter Arten von Injektions-/Infusionssets adsorbiert werden. In diesem Fall könnte es zu einem Versagen der Therapie kommen. Aus diesem Grund dürfen für FEIBA nur zugelassene Injektions-/Infusionssets aus Kunststoff verwendet werden.
6.3 Dauer der Haltbarkeit
2 Jahre
Die chemische und physikalische Stabilität des rekonstituierten Produktes ist über 3 Stunden bei 20 °C -25 °C belegt.
Aus mikrobiologischer Sicht sollte es jedoch unmittelbar verbraucht werden. Wenn dies nicht geschieht, ist der Anwender für die Lagerungsbedingungen und die Lagerungszeit verantwortlich. Die gebrauchsfertige Lösung darf nicht gekühlt werden.
Nach Ablauf des Verfalldatums darf FEIBA nicht mehr angewendet werden.
6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Nicht über 25 °C lagern. Nicht einfrieren.
Arzneimittel in der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen.
6.5 Art und Inhalt des Behältnisses
Das Pulver befindet sich in einer Durchstechflasche aus farblosem Glas der hydrolytischen Klasse II, das Lösungsmittel in einer Durchstechflasche aus farblosem Glas der hydrolytischen Klasse I. Beide Durchstechflaschen sind mit Butylgummistopfen und Schutzkappen verschlossen.
Packungsinhalt:
oder 1
1 Durchstechflasche FEIBA NF 500 E oder FEIBA NF 1000 E 1 Durchstechflasche mit 20 ml Wasser für Injektionszwecke 1 Einmalspritze 1 Einmalnadel
1 Butterflynadel mit Klammer (Flügelkanüle zur Injektion)
1 Filternadel 1 Transfernadel 1 Belüftungsnadel
Durchstechflasche FEIBA NF 500 E oder FEIBA NF 1000 E 1 Durchstechflasche mit 20 ml Wasser für Injektionszwecke 1 BAXJECT II Hi-Flow 1 Einmalspritze 1 Einmalnadel
1 Butterflynadel mit Klammer (Flügelkanüle zur Injektion)
6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung
FEIBA nur mit dem Wasser für Injektionszwecke und den beigepackten Medizinprodukten auflösen. Während der gesamten Handhabung auf aseptische Arbeitsweise achten.
FEIBA erst unmittelbar vor der Verabreichung auflösen. Das FEIBA-Pulver muss vollständig gelöst sein. Die gebrauchsfertige Lösung sofort verwenden (da das Produkt keine Konservierungsmittel enthält).
Die Lösung vor der Anwendung auf sichtbare Partikel und Verfärbung prüfen. Die Lösung ist klar oder leicht opaleszierend. Lösungen, die trüb sind oder Ablagerungen aufweisen, nicht verwenden.
Das nadellose Transfersystem oder die Überleitungsnadel nicht verwenden, wenn die sterilen Systeme oder ihre Verpackungen beschädigt sind oder Anzeichen der Manipulation aufweisen.
Wenn andere Medizinprodukte als die beigepackten zusammen mit FEIBA verwendet werden, ist sicherzustellen, dass ein entsprechender Filter mit einer Porengröße von mindestens 149 gm verwendet wird.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Auflösen des Pulvers mit dem BAXJECT II Hi-Flow:
1. Falls erforderlich, das ungeöffnete Lösungsmittelfläschchen (Wasser für Injektionszwecke) auf Raumtemperatur bringen, z. B. mit Hilfe eines sterilen Wasserbades zum Erwärmen innerhalb von wenigen Minuten (max. +37 °C).
2. Die Schutzkappen von den Flaschen mit Konzentrat und Lösungsmittel (Abb. 1) entfernen und die Gummistopfen beider Flaschen reinigen.
3. Die Verpackung des BAXJECT II Hi-Flow öffnen, indem die Schutzfolie abgezogen wird ohne dabei den Packungsinhalt zu berühren (Abb. a). Das Transfersystem nicht aus der Verpackung nehmen.
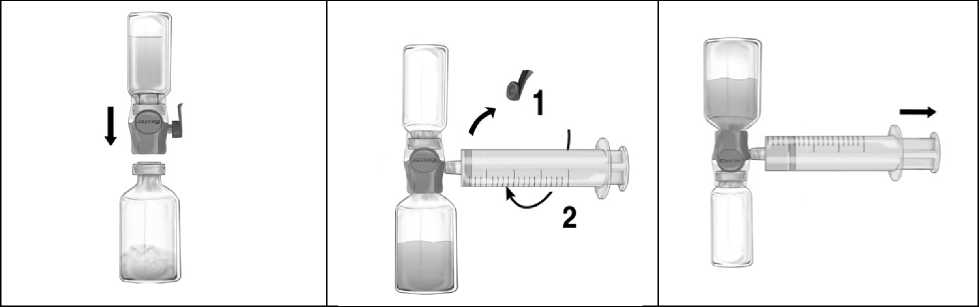
4. Die Öffnung nach unten drehen und den durchsichtigen Plastikdorn durch den Gummistopfen der Lösungsmittelflasche drücken (Abb. b). Nun die Verpackung vom BAXJECT II Hi-Flow am Ende fassend abnehmen (Abb. c). Die blaue Schutzkappe noch nicht vom BAXJECT II Hi-Flow entfernen.
5. Das System, bestehend aus dem BAXJECT II Hi-Flow und der Lösungsmitteldurchstechflasche, nun wenden, so dass sich die Lösungsmittelflasche oben befindet. Den purpurfarbenen Dorn des BAXJECT II Hi-Flow durch den Gummistopfen der FEIBA -Flasche drücken. Durch das Vakuum wird das Lösungsmittel in die FEIBA -Flasche gezogen (Abb. d).
6. Vorsichtig schwenken, NICHT schütteln, bis das gesamte Pulver vollständig gelöst ist. Sicherstellen, dass FEIBA vollständig gelöst ist, da sonst ein Teil des FEIBA-Wirkstoffs im Filter des Transfersystems zurückgehalten wird.
Abb. a

Abb. b
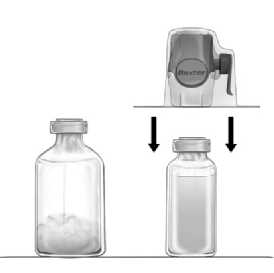
Abb. c

|
Abb. d |
Abb. e |
Abb. f |
Hinweise zur Injektion/Infusion
1. Die blaue Schutzkappe vom BAXJECT II Hi-Flow entfernen. Die Spritze fest an den BAXJECT II Hi-Flow anschließen KEINE LUFT IN DIE SPRITZE AUFZIEHEN! (Abbe). Um eine feste Verbindung zwischen der Spritze und dem BAXJECT II Hi-Flow zu gewährleisten, wird der Gebrauch einer Luer-Lock Spritze empfohlen (die Spritze beim Anschließen im Uhrzeigersinn drehen bis ein Widerstand zu spüren ist),
2. Das System umdrehen, so dass sich das Fläschchen mit dem gelösten Produkt oben befindet. Die FEIBA -Lösung durch LANGSAMES Zurückziehen des Kolbens in die Spritze aufziehen (Abb.f). Stellen Sie sicher, dass die feste Verbindung zwischen Spritze und BAXJECT II Hi-Flow während des gesamten Aufzieh-Vorgangs erhalten bleibt.
3. Die Spritze entfernen.
4. Falls die Lösung in der Spritze aufgeschäumt ist, warten bis der Schaum zusammen gefallen ist. Die Lösung mit der beigepackten Flügelkanüle zur Injektion (oder der Einmalnadel) intravenös injizieren.
Injektions-/Infusionsrate von 2 Einheiten FEIBA pro kg Körpergewicht in der Minute nicht
überschreiten.
Auflösen des Pulvers mit dem Nadelset:
1. Falls erforderlich, das ungeöffnete Lösungsmittelfläschchen (Wasser für Injektionszwecke) auf Raumtemperatur bringen, z. B. mit Hilfe eines sterilen Wasserbades zum Erwärmen innerhalb von wenigen Minuten (max. +37 °C).
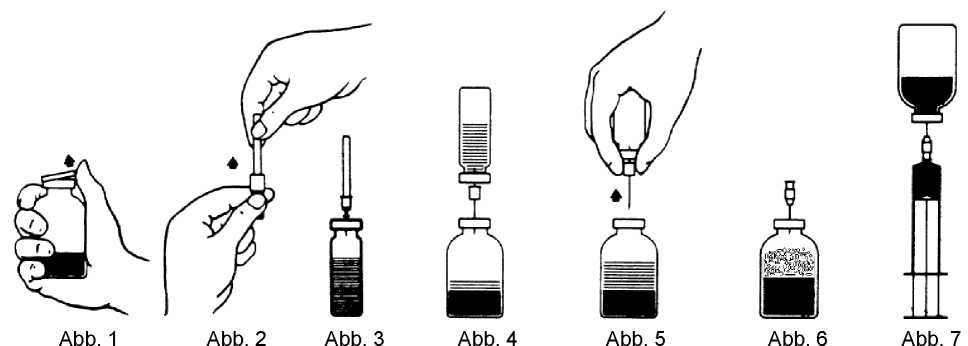
2. Die Schutzkappen von den Flaschen mit Konzentrat und Lösungsmittel (Abb. 1) entfernen und die Gummistopfen beider Flaschen reinigen.
3. Die Schutzkappe von einem Ende der mitgelieferten "Transfernadel" durch Drehen und Ziehen entfernen (Abb. 2). Die freigelegte Nadel durch den Gummistopfen der Lösungsmittelflasche stechen (Abb. 3).
4. Die Schutzkappe von der anderen Seite der Transfernadel abziehen, ohne das freie Ende zu berühren.
5. Die Lösungsmittelflasche kopfüber über die Konzentratflasche halten und das freie Ende der Transfernadel durch den Gummistopfen in der Konzentratflasche stechen (Abb. 4). Durch das entstehende Vakuum wird das Lösungsmittel angesaugt.
6. Die beiden Flaschen trennen, indem Sie die Transfernadel von der Konzentratflasche entfernen (Abb. 5). Den Lösungsvorgang durch sanftes und gleichmäßiges Schwenken der Konzentratflasche beschleunigen.
7. Ist das Konzentrat vollständig aufgelöst, die mitgelieferte "Belüftungsnadel" (Abb. 6) einführen, so dass eventuell vorhandener Schaum zusammenfällt. Anschließend die Belüftungsnadel wieder entfernen.
Hinweise zur Injektion/Infusion:
1. Die Schutzkappe der mitgelieferten "Filtemadel" durch Drehen und Ziehen entfernen und die Nadel auf eine Einmalspritze setzen. Die Lösung in die Spritze aufziehen (Abb. 7).
2. Die Filternadel von der Spritze trennen und die Lösung mit Hilfe der mitgelieferten "Flügelkanüle" (oder der mitgelieferten Einmalnadel) langsam intravenös applizieren.
Bei Verabreichung durch Infusion, ein Einmal-Infusionsset mit passendem Filter verwenden.
Injektions-/Infusionsrate von 2 Einheiten FEIBA pro kg Körpergewicht in der Minute nicht überschreiten.
7. INHABER DER ZULASSUNG
Baxter Deutschland GmbH Edisonstraße 4 85716 Unterschleißheim Telefon: 089/31701-0 Fax: 089/31701-177 E-Mail: info_de@baxter.com
8. ZULASSUNGSNUMMERN
PEI.H.03340.01.1
PEI.H.03340.02.1
9. DATUM DER ERTEILUNG DER ZULASSUNG/VERLÄNGERUNG DER ZULASSUNG
6. Dezember 2005
10. STAND DER INFORMATION
September 2014
11. SONSTIGE HINWEISE
Herkunftsländer der zur Produktion verwendeten Plasmen: Deutschland, Finnland, Norwegen, Österreich, Schweden, Schweiz, Tschechien und Vereinigten Staaten von Amerika.
12. VERKAUFSABGRENZUNG
Verschreibungspflichtig
a
b
c
Eine präzise Schätzung der Inzidenz dieser Nebenwirkungen ist auf der Grundlage der verfügbaren Daten nicht möglich.
„Anstieg des Hemmkörper-Titers (anamnestische Reaktion)“ [kein Preferred Term gemäß MedDRA] bedeutet ein Anstieg eines vorbestehenden Hemmkörper-Titers im Anschluss an die Gabe von FEIBA. Siehe Abschnitt 4.4.
Nebenwirkungen, die in den ursprünglichen klinischen Studien und in der Prophylaxe-Studie berichtet wurden. Die Häufigkeiten ergeben sich aus der Studie zur Prophylaxe.
Nebenwirkungen, die in der Studie zur Prophylaxe berichtet wurden. Die Angaben zu den Häufigkeiten beziehen sich nur auf diese Studie.
Seite 15 von 15