Gammagard S/D
□□□□□□
Fachinformation
1. BEZEICHNUNG DES ARZNEIMITTELS Gammagard S/D
2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
Normales Immunglobulin G vom Menschen (IgG)
Gammagard S/D liegt als Pulver und Lösungsmittel zur Infusion vor, und enthält in der Durchstechflasche (0,5 g; 2,5 g; 5,0 g; 10,0 g) normales Immunglobulin vom Menschen (IgG). Gammagard S/D ist mit Sterilisiertem Wasser für Injektionszwecke zu einer 5%igen Lösung (50 mg/ml) oder zu einer 10%igen Proteinlösung (100 mg/ml) zu rekonstituieren mit einem IgG-Gehalt von mindestens 90 %.
Verteilung der Subklassen:
|
IgG1 |
> 56,9 % |
|
IgG2 |
> 16,0 % |
|
IgG3 |
> 3,3 % |
|
IgG4 |
> 0,3 % |
Maximaler IgA-Gehalt: <3 Mikrogramm pro ml in einer 5%igen Lösung.
Hergestellt aus menschlichem Plasma
Sonstige Bestandteile mit bekannter Wirkung: Natriumchlorid, Glukosemonohydrat (siehe 4.4). Die vollständige Auflistung der sonstigen Bestandteile siehe Abschnitt 6.1.
3. DARREICHUNGSFORM
Pulver und Lösungsmittel zur Herstellung einer Infusionslösung.
Gammagard S/D ist ein lyophilisiertes, weißes oder schwach gelbliches Pulver/Pellet, frei von sichtbaren, fremden Partikeln.
4. KLINISCHE ANGABEN
4.1. Anwendungsgebiete
Substitutionsbehandlung an Erwachsenen, Kindern und Jugendlichen (0-18 Jahre) bei:
• primären Immundefekten (PID) mit verminderter Antikörperproduktion (siehe Abschnitt 4.4.)
• Hypogammaglobulinämie und rezidivierenden, bakteriellen Infekten bei Patienten mit chronisch lymphatischer Leukämie, wenn eine prophylaktische Antibiotikabehandlung fehlgeschlagen ist.
• Hypogammaglobulinämie und rezidivierenden, bakteriellen Infekten bei Patienten in der Plateauphase eines multiplen Myeloms, wenn die Immunantwort auf eine Pneumokokkenimpfung ausgeblieben ist.
• Hypogammaglobulinämie bei Patienten nach allogenen, hämatopoetischen StammzellTransplantationen (HSCT)
• kongenitaler HIV-Infektion (AIDS) und rezidivierenden Infekten.
Immunmodulation an Erwachsenen, Kindern und Jugendlichen (0-18 Jahre) mit:
• Primärer Immunthrombozyopenie (Idiopathische thrombozytopenische Purpura [ITP]) bei Patienten mit hohem Blutungsrisiko oder vor Operationen zur Korrektur der Thrombozytenzahl
• Guillain-Barre-Syndrom
• Kawasaki Syndrom
4.2. Dosierung, Art und Dauer der Anwendung
Beginn und Überwachung der Therapie sollten unter der Aufsicht eines mit der Behandlung von Immunmangelkrankheiten erfahrenen Arztes erfolgen.
Dosierung
Dosierung und Dosierungsintervalle sind abhängig von der Indikation.
Bei Substitutionsbehandlungen kann eine individuelle Dosierung für jeden Patienten in Abhängigkeit von der pharmakokinetischen und klinischen Reaktion notwendig sein. Folgende Dosierungsangaben können als Richtlinie gelten.
Substitutionsbehandlung bei primären Immundefekten:
Bei der Dosierung sollte ein IgG-Plasmaspiegel von mindestens 5-6 g/l angestrebt werden (gemessen vor der nächsten Infusion). Nach Behandlungsbeginn werden 3-6 Monate benötigt, um ein Gleichgewicht einzustellen. Die empfohlene Initialdosis beträgt 0,4-0,8 g/kg Körpergewicht (KG) gefolgt von 0,2 g/kg KG alle 3-4 Wochen.
Um einen Talspiegel von 5-6 g/l zu erreichen, ist eine Erhaltungsdosis von 0,2-0,8 g/kg KG pro Monat erforderlich. Die Dosierungsintervalle können bei Vorliegen eines konstanten Plasmaspiegels („steady state“) 3-4 Wochen betragen.
Um die Dosierung und Dosierungsintervalle individuell anpassen zu können, sollten die Plasmatalspiegel regelmäßig kontrolliert und das Neuauftreten von Infektionen überwacht werden. Um die Infektionsrate zu reduzieren und um einen höheren Plasmaspiegel zu erreichen, kann es notwendig werden die Dosierung zu steigern.
Hypogammaglobulinämie und rezidivierende, bakterielle Infekte bei Patienten mit chronisch lymphatischer Leukämie, wenn eine prophylaktische Antibiotikabehandlung fehlgeschlagen ist; Hypogammaglobulinämie und rezidivierende, bakterielle Infekte bei Patienten in der Plateauphase eines multiplen Myeloms, wenn die Immunantwort auf eine Pneumokokkenimpfung ausgeblieben ist; Kinder und Jugendliche mit kongenitaler HIV-Infektion (AIDS) und rezidivierenden Infekten.
Die empfohlene Dosis beträgt 0,2-0,4 g/kg alle 3-4 Wochen.
Hypogammaglobulinämie bei Patienten nach allogener, hämatopoetischer Stammzell-Transplantation: Die empfohlene Dosis beträgt 0,2-0,4 g/kg alle 3-4 Wochen. Der Plasmatalspiegel sollte oberhalb von 5 g/l gehalten werden.
Primäre Immunthrombozytopenie:
Es gibt 2 unterschiedliche Behandlungsschemata
• 0,8-1 g/kg am 1. Tag, gefolgt von der gleichen Dosis innerhalb von 3 Tagen
• 0,4 g/kg täglich für 2-5 Tage
Die Behandlung kann bei einem Rückfall wiederholt werden.
Guillain-Barre-Syndrom:
0,4 g/kg an 5 aufeinanderfolgenden Tagen.
Kawasaki-Svndrom:
1,6-2,0 g/kg sollten auf mehrere Dosen verteilt über 2-5 Tage gegeben werden; oder 2 g/kg als Einzeldosis. Die Patienten sollten gleichzeitig mit Acetylsalicylsäure behandelt werden.
Die Dosierungsempfehlungen werden in folgender Tabelle zusammengefasst:
|
Indikation |
Dosierung |
Häufigkeit der Verabreichung |
|
Substitutionstherapie bei primären Immundefekten |
- Initialdosis: 0,4-0,8 g/kg KG |
alle 3-4 Wochen, um die IgG-Plasmatalspiegel auf mindestens 5-6 g/l zu halten |
|
- anschließend: 0,2-0,8 g/kg KG | ||
|
Substitutionstherapie bei sekundären Immundefekten |
0,2-0,4 g/kg KG |
alle 3-4 Wochen, um die IgG-Plasmatalspiegel auf mindestens 5-6 g/l zu halten |
|
kongenitale HIV-Infektion (AIDS) |
0,2-0,4 g/kg KG |
alle 3-4 Wochen |
|
Hypogammaglobulinämie (<4 g/l) bei Patienten nach allogener, hämatopoetischer StammzellTransplantation) |
0.2 -0.4 g/kg |
alle 3-4 Wochen, um die IgG-Plasmatalspiegel oberhalb von 5 g/l zu halten |
|
Immunmodulation: | ||
|
Primäre Immunthrombozytopenie |
0,8-1 g/kg KG oder |
am 1. Tag, gefolgt von der gleichen Dosis innerhalb von 3 Tagen, falls erforderlich |
|
Guillain-Barre-Syndrom |
0,4 g/kg KG/Tg |
für 2-5 Tage |
|
0,4 g/kg KG |
für 5 Tage | |
|
Kawasaki-Syndrom |
1,6-2 g/kg KG oder |
mehrere Dosen über 2-5 Tage verteilt, zusammen mit Acetylsalicylsäure |
|
2 g/kg KG |
als Einzeldosis zusammen mit Acetylsalicylsäure |
Pädiatrische Population:
Die Dosierung an Kindern und Jugendlichen (0-18 Jahre) unterscheidet sich nicht von den Erwachsenen, da die Dosierung für jede Indikation nach Körpergewicht erfolgt und gemäß den o. g. Angaben dem klinischen Erfolg angepasst wird.
Art der Anwendung
Zur intravenösen Anwendung
Es wird empfohlen zur Applikation der 10%igen Gammagard S/D-Lösung möglichst die Ellbogenvenen zu verwenden. Dies kann die Wahrscheinlichkeit für das Auftreten von Beschwerden an der Infusionsstelle für den Patienten reduzieren.
5%iges Gammagard S/D (50 mg/ml) sollte mit einer Anfangsgeschwindigkeit von 0,5 ml/kg Körpergewicht und Stunde intravenös verabreicht werden. Generell wird empfohlen, dass bei Patienten, die eine Behandlung mit Gammagard S/D beginnen, oder von einem intravenösen Immunglobulin G auf Gammagard S/D umgestellt werden, mit der geringstmöglichen Infusionsgeschwindigkeit begonnen wird. Wenn die Patienten einige Infusionen mittlerer Infusionsgeschwindigkeit vertragen haben, wird diese auf die maximale Infusionsgeschwindigkeit angehoben (siehe Abschnitt 4.4). Bei guter Verträglichkeit kann die Infusionsgeschwindigkeit langsam bis auf maximal 4 ml/kg Körpergewicht und Stunde gesteigert werden.
Patienten, die Gammagard S/D als 5%ige Lösung mit 4 ml/kg KG und Stunde gut vertragen, können mit der 10%igen Konzentration, beginnend mit 0,5 ml/kg KG und Stunde, behandelt werden. Sollten keine Nebenwirkungen auftreten, kann die Infusionsgeschwindigkeit schrittweise auf eine maximale Rate von 8 ml/kg KG und Stunde gesteigert werden.
Bei Patienten mit dem Risiko eines akuten Nierenversagens oder thromboembolischen Ereignissen sollen IVIG-Präparate mit der geringstmöglichen Infusionsgeschwindigkeit und in der kleinstmöglichen Dosierung verabreicht werden.
4.3. Gegenanzeigen
Überempfindlichkeit oder bekannte anaphylaktische Reaktionen gegen den Wirkstoff oder einen der sonstigen Bestandteile (siehe Abschnitt 4.4.).
Überempfindlichkeit gegen humane Immunglobuline, insbesondere bei Patienten mit Antikörpern gegen
IgA.
4.4. Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Eine 5%ige Gammagard S/D-Lösung enthält 5 g Glukose pro 100 ml Lösung oder 5 g Glukose pro 5 g IgG, d. h. ein 70 kg schwerer Patient, der mit bei einer Dosis von 1 g/kg KG Gammagard S/D behandelt wird, würde 70 g Glukose erhalten (das entspräche 280 Kalorien). Dies sollte bei latentem Diabetes (wobei eine vorübergehende Glukosurie auftreten kann), bei Diabetes oder bei Patienten mit einer zuckerreduzierten Diät berücksichtigt werden. Zum akuten Nierenversagen die unten genannten Angaben beachten.
Bestimmte schwere Unverträglichkeitsreaktionen können mit der Infusionsgeschwindigkeit zusammenhängen. Die in Abschnitt 4.2. "Dosierung, Art und Dauer der Anwendung” empfohlene Infusionsgeschwindigkeit sollte daher genau befolgt werden. Die Patienten sind während der gesamten Infusionsdauer genau zu überwachen und in Hinblick auf eventuell auftretende Symptome zu beobachten. Bestimmte Nebenwirkungen können häufiger auftreten bei:
• hoher Infusionsgeschwindigkeit
• Patienten, die normales Immunglobulin vom Menschen das erste Mal erhalten, oder in seltenen Fällen, bei einem Präparatewechsel, oder wenn die Behandlung für längere Zeit unterbrochen wurde.
Mögliche Komplikationen können oft vermieden werden, wenn sichergestellt wird, dass Patienten:
• nicht gegen normales Immunglobulin vom Menschen überempfindlich sind, indem das Produkt erstmalig sehr langsam infundiert wird (0,5-1 ml/kg KG/Stunde)
• während der Infusionsdauer sorgfältig auf etwaige Symptome hin überwacht werden. Insbesondere sollten Patienten, die das erste Mal Immunglobulin vom Menschen erhalten, Patienten die sich einem bei Präparatewechsel unterziehen, oder die eine längere Therapieunterbrechung hatten, sorgfältig für die Dauer der Erstinfusion und während der ersten Stunde nach der Erstinfusion überwacht werden, um mögliche Nebenwirkungen zu bemerken. Alle anderen Patienten sollten nach der Verabreichung mindestens 20 Minuten unter Beobachtung bleiben;
• sichergestellt ist, dass der Glukose-Gehalt (maximal 0,4 g/g IgG) bei einer latenten Störung im Glukosehaushalt (wobei eine vorübergehende Glukosurie auftreten kann), bei Diabetes oder bei Patienten mit einer zucker-reduzierten Diät berücksichtigt wird.
Bei Unverträglichkeitsreaktionen ist entweder die Infusionsgeschwindigkeit zu verringern oder die Infusion zu unterbrechen. Die benötigte Behandlung ist von der Art und Schwere der Nebenwirkungen abhängig. Bei Auftreten von Schocksymptomen sollte die Behandlung nach den Regeln der Schocktherapie erfolgen.
Alle Patienten, die intravenöse Immunglobuline erhalten, benötigen:
• eine adäquate Hydratation vor Beginn der intravenösen Immunglobulintherapie
• eine Überwachung der Harnproduktion
• eine Überwachung der Serum-Kreatininspiegel
• die Vermeidung einer Begleittherapie mit Schleifendiuretika
Überempfindlichkeit
Echte Überempfindlichkeitsreaktionen sind selten. Sie können in den sehr seltenen Fällen von IgA-Mangel mit anti IgA-Antikörpern auftreten.
Intravenöses Immunglobulin ist nicht indiziert bei Patienten mit selektivem IgA-Mangel, wenn der IgA-Mangel die einzige immunologische Abnormität darstellt.
Selten kann normales Immunglobulin vom Menschen eine anaphylaktische Reaktion mit Blutdruckabfall hervorrufen, sogar bei Patienten, die die Behandlung bisher gut vertragen haben.
Thromboembolie
Ein klinischer Zusammenhang zwischen der Behandlung mit intravenösem Immunglobulin (IVIG) und thromboembolischen Komplikationen wie Herzinfarkt, zerebrovaskuläre Ereignisse (wie Schlaganfall), Lungenembolie und tiefen Venenthrombosen wurde nachgewiesen. Dies ist vermutlich zurückzuführen auf eine relative Erhöhung der Blutviskosität durch den hohen Einstrom von Immunglobulin bei Risikopatienten. Besondere Vorsicht ist unbedingt erforderlich bei der Verschreibung und Infusion von IVIG an übergewichtige Patienten und an Patienten mit vorbestehenden Risikofaktoren für thrombotische Ereignisse wie Arteriosklerose, multiple kardiogene Risikofaktoren, fortgeschrittenes Alter, verminderte Herzleistung, vermutete Hyperviskosität wie z. B. Dehydratation oder Paraproteinämie, Hyperkoagulabilität, bei Patienten mit längeren Phasen einer Immobilität, mit Übergewicht, bei Patienten denen Östrogene verabreicht werden, bei Patienten mit Diabetes mellitus, mit erworbenen oder angeborenen thrombophilen Erkrankungen, sowie bei Patienten mit einer Anamnese mit Gefäßerkrankungen, thrombotischen Ereignissen oder bei Patienten mit liegendem Gefäßkatheder.
Bei Patienten mit dem Risiko thromboembolischer Nebenwirkungen sollte Gammagard S/D mit der geringstmöglichen Infusionsgeschwindigkeit und der kleinstmöglichen Dosis verabreicht werden.
Akutes Nierenversagen
Fälle von akutem Nierenversagen wurden bei Patienten beschrieben, die eine Therapie mit IVIg erhielten. In den meisten Fällen wurden Risikofaktoren erkannt, z. B. vorbestehende Niereninsuffizienz, Diabetes mellitus, Hypovolämie, Übergewicht, nephrotoxische Begleitmedikation oder Alter über 65 Jahre.
Im Falle einer Beeinträchtigung der Nierenfunktion sollte ein Absetzen des IVIg-Präparates erwogen werden. Die Berichte über Nierenfunktionsstörungen und akutes Nierenversagen wurden zwar mit der Anwendung vieler zugelassener IVIg-Präparate mit verschiedenen sonstigen Bestandteilen wie Saccharose, Glucose und Maltose, in Verbindung gebracht, jedoch war der Anteil der Präparate mit Saccharose als Stabilisator unverhältnismäßig hoch. Bei Risikopatienten kann die Anwendung von IVIg-Präparaten ohne diese sonstigen Bestandteile erwogen werden. Gammagard S/D enthält keine Saccharose oder Maltose.
IVIg-Präparate sollten bei Patienten, bei denen ein Risiko für akutes Nierenversagen besteht, mit möglichst geringer Infüsionsgeschwindigkeit und in möglichst niedriger Dosierung verabreicht werden.
Transfusionsbedingte akute Lungenverletzung (TRALI-Syndrom)
Es wurde von non-kardiogenen Pulmonalödemen (transfusionsbedingte akute Lungenverletzung, TRALI-Syndrom) bei Patienten berichtet, denen IVIg verabreicht wurde.
Aseptisches Meningitis-Svndrom
Ein aseptisches Meningitis-Syndrom (AMS) wurde im Zusammenhang mit IVIG (einschließlich Gammagard S/D) berichtet. Ein Abbruch der intravenösen Immunglobulintherapie kann zu einer Remission innerhalb von einigen Tagen führen. Die Symptome beginnen normalerweise innerhalb weniger Stunden bis 2 Tagen nach Beginn der IVIG-Behandlung.
• Untersuchungen am Liquor zeigen häufig eine Pleozytose bis zu mehreren Tausend Zellen pro mm3. Dabei handelt es sich überwiegend um Granulozyten und gleichzeitig erhöhte Proteinwerte bis zu mehreren Hundert mg/dl.
• AMS tritt häufig bei hohen Dosen der IVIG (2 g/kg) auf.
Hämolytische Anämie
Gammagard S/D enthält Blutgruppen-Antikörper, die als Hämolysine wirken und in vivo die Beladung der roten Blutkörperchen mit Immunglobulinen (RBC) veranlassen können. Diese Antikörper können eine positive, direkte Antiglobulin-Reaktion (positiver Coombs-Test) verursachen. Im Anschluss an eine Therapie mit Gammagard S/D kann sich durch eine beschleunigte RBC-Clearance eine verzögerte, hämolytische Anämie entwickeln, Es wurde auch über akute Hämolysen in Übereinstimmung mit intravaskulären Hämolysen berichtet.
Die folgenden Risikofaktoren können im Zusammenhang mit der Entwicklung einer Hämolyse stehen: hohe Dosen (als Einzelanwendung oder verteilt über mehrere Tage) und andere Blutgruppen als Blutgruppe 0. Im Einzelfall kann eine entzündliche Vorerkrankung das Risiko einer Hämolyse erhöhen, auch wenn der Zusammenhang unklar ist.
Weitere Vorsichtsmaßnahmen
Bei Patienten unter IVIG-Behandlung können Hyperproteinämie und erhöhte Serumviskosität auftreten. Vor der Verabreichung von Gammagard S/D sollte bei den Patienten auf eine adäquate Hydratation geachtet werden. Patienten mit dem Risiko einer Hyperviskosität sollten auf Anzeichen und Symptome einer Thrombose und auf eine erhöhte Blutviskosität hin überwacht werden.
Die enthaltene Natriummenge muss hinsichtlich der maximalen Tagesdosis bei Patienten, die unter einer natriumkontrollierten Diät stehen in Bezug zur empfohlenen Tagesmenge gesetzt werden. Bei diesen Patienten sollte die im Produkt enthaltene Natriummenge errechnet und bei der Bestimmung des diätetischen Natriums berücksichtigt werden. Eine 5%ige Gammagard S/D-Lösung enthält etwa 0,85 % Natriumchlorid oder etwa 67,8 mg Natrium pro Gramm IgG, d. h. ein 70 kg schwerer Patient, der mit 1 g/kg KG Gammagard S/D behandelt wird, würde 4676 mg Natrium erhalten.
Übertragbare Erreger
Gammagard S/D wird aus humanem Plasma hergestellt. Standardmaßnahmen zur Vorbeugung von Infektionen, die sich durch den Einsatz von Arzneimitteln ergeben, die aus Blut oder Blutplasma hergestellt sind, schließen die Auswahl der Spender und das Screening jeder Einzelspende und jedes Plasmapools auf spezifische Infektionsmarker sowie effektive Schritte zur Inaktivierung/Entfernung von Viren im Herstellungsverfahren ein. Dennoch kann bei der Verabreichung von Arzneimitteln aus menschlichem Blut oder Blutplasma die Möglichkeit der Übertragung von Krankheitserregern nicht völlig ausgeschlossen werden. Dasselbe gilt auch für bislang unbekannte oder neu aufgetretene Viren und andere Pathogene.
Die durchgeführten Maßnahmen werden als wirksam gegen umhüllte Viren wie das human Immunschwächevirus (HIV), das Hepatitis B-Virus (HBV), das Hepatitis C-Virus (HCV) und gegen nicht-umhüllte Viren wie Hepatitis A-Virus (HAV) und Parvovirus B19 erachtet.
Die klinischen Erfahrungen zeigen ein geringes Risiko hinsichtlich einer Übertragung von Hepatitis A oder Parvovirus B19 durch Immunoglobuline; man geht auch davon aus, dass der Antikörpergehalt einen wesentlichen Beitrag zur Virensicherheit leistet.
Auswirkungen auf serologische Untersuchungen
Nach Infusion von Immunglobulinen kann es durch den vorübergehenden Anstieg der verschiedenen, passiv übertragenen Antikörper im Blut des Patienten zu falschen Testergebnissen bei serologischen Untersuchungen wie z. B. Hepatitis A, Hepatitis B, Masern und Windpocken kommen.
Die passive Übertragung von Antikörpern gegen Erythrozytenantigene, wie A, B und D kann einige serologische Untersuchungen auf Erythrozyten-Antikörper wie z. B. den Antiglobulintest (DAT, Coombs-Test) beeinträchtigen.
Es wird auf die Dokumentationspflicht gemäß Transfüsionsgesetz hingewiesen.
Es wird dringend empfohlen, jedes Mal, wenn einem Patienten Gammagard S/D verabreicht wird, den Namen des Patienten und die Chargennummer des Arzneimittels zu notieren, um genau nachverfolgen zu können, welcher Patient mit Arzneimittel einer bestimmten Charge behandelt wurde.
Pädiatrische Population
Die besonderen Warnhinweise und Vorsichtsmaßnahmen für Erwachsene sollten auch bei Kindern und Jugendlichen beachtet werden.
4.5. Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Virus-Lebendimpfstoffe
Die Verabreichung von Immunglobulinen kann die Wirkung von Virus-Lebendimpfstoffen wie Masern, Röteln, Mumps und Varizellen über einen Zeitraum von mindestens 6 Wochen bis zu 3 Monaten beeinträchtigen. Nach Verabreichung des Produkts, soll daher ein Zeitraum von 3 Monaten verstreichen, bevor eine Impfung mit einem Virus-Lebendimpfstoff erfolgt. Bei Masern kann dieser Zeitraum bis zu 1 Jahr andauern. Deshalb sollte bei Patienten, die eine Masernimpfung erhalten haben, der Antikörperspiegel überprüft werden.
Pädiatrische Population:
Es ist davon auszugehen, dass die für Erwachsene aufgeführten Wechselwirkungen auch bei Kindern und Jugendlichen auftreten können.
4.6. Fertilität, Schwangerschaft und Stillzeit
Schwangerschaft
Die Sicherheit von Gammagard S/D während der Schwangerschaft wurde nicht untersucht. Daher sollte Gammagard S/D nur mit Vorsicht schwangeren oder stillenden Frauen gegeben werden. Es konnte gezeigt werden, dass IVIG, die der Mutter verabreicht werden, ab dem dritten Schwangerschaftsdrittel zunehmend durch die Plazenta übertreten. Die Klinische Erfahrung mit Immunglobulinen weist darauf hin, dass keine schädlichen Wirkungen auf den Verlauf der Schwangerschaft, den Fötus und das Neugeborene zu erwarten
Stillzeit
Immunglobuline werden über die Muttermilch auf den Säugling übertragen und tragen so dazu bei das Neugeborene vor Pathogenen zu schützen, die über die Mukosa eindringen.
Fertilität
Die Auswirkungen von Gammagard S/D auf die Fertilität wurde nicht in klinischen Prüfungen untersucht
4.7. Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen können durch einige Nebenwirkungsreaktionen von Gammagard S/D beeinträchtigt sein. Patienten, die während der Behandlung Nebenwirkungsreaktionen bemerken, sollten bis zu deren Abklingen nicht am Verkehr teilnehmen oder Maschinen bedienen.
4.8. Nebenwirkungen
4.8.1. Nebenwirkungen aus Klinischen Prüfungen
Die Nebenwirkungen aus einer pivotalen klinischen Studie von Gammagard S/D sowie einer Phase 4 Studie zur Erfassung der akuten und mittelfristigen Sicherheit von Gammagard S/D wurden zusammengefasst.
|
Nebenwirkungen von Gammagard S/D | ||
|
Systemorganklassen gemäß MedDRA |
Bevorzugter Begriff in der MedDRA- |
Häufigkeit der ADR* |
|
Infektionen und parasitäre Erkrankungen |
Influenza |
gelegentlich |
|
aseptische Meningitis |
unbekannt | |
|
Erkrankungen des Blutes und des Lymphsystems |
Hämolysen, Anämie, Thrombozytopenie, Lymphade-nopathie |
unbekannt |
|
Stoffwechsel- und Ernährungsstörungen |
verminderter Appetit |
gelegentlich |
|
Erkrankungen des Immunsystems |
anaphylaktische oder anaphylaktoide Reaktion, anaphylaktischer Schock, Überempfindlichkeit |
unbekannt |
|
Psychiatrische Erkrankungen |
Angstanfälle |
gelegentlich |
|
Agitation |
unbekannt | |
|
Ruhelosigkeit |
gelegentlich | |
|
Erkrankungen des Nervensystem |
Kopfschmerzen |
häufig |
|
Lethargie |
gelegentlich | |
|
Zerebrovaskuläre Anfälle, Schlaganfälle, vorübergehende ischämische Attacken, Krämpfe, Migräne, Benommenheit, Parästhesien, Synkopen, Tremor, Schwindel |
unbekannt | |
|
Augenerkrankungen |
verschwommenes Sehen |
gelegentlich |
|
Retinale Venenthrombose, Augenschmerzen, Photophobie |
unbekannt | |
|
Herzerkrankungen |
Herzklopfen |
gelegentlich |
|
Herzinfarkt, Zyanose, Tachykardie, Bradykardie |
unbekannt | |
|
Gefäßerkrankungen |
Flush |
häufig |
|
Blutdruckschwankungen |
gelegentlich | |
|
Arterielle Thrombose, Thrombose der Vena cava, tiefe Venenthrombose, Thrombophlebitis, Hypotension, Hypertension, Blässe |
unbekannt | |
|
Erkrankungen der Atemwege, des Brustraums und des Mediastinums |
Atemnot, Nasenbluten |
gelegentlich |
|
Lungenembolie, Lungenödem, Hypoxie, Bronchospasmus, keuchende Atmung, Hyperventilation, Engegefühl im Hals, Husten |
unbekannt | |
|
Erkrankungen des Gastrointestinaltrakts |
Erbrechen, Übelkeit |
häufig |
|
Durchfall, Stomatitis, Schmerzen und Unwohlsein im oberen Bauchraum |
gelegentlich | |
|
Bauchschmerzen, Verdauungsstörungen |
unbekannt | |
|
Leber-und Gallenerkrankungen |
nicht-infektiöse Hepatitis |
unbekannt |
|
Erkrankungen der Haut |
Urtikaria, Pruritus, kalter Schweiß, Hyperhidrosis |
gelegentlich |
|
Nebenwirkungen von Gammagard S/D | ||
|
Systemorganklassen gemäß MedDRA |
Bevorzugter Begriff in der MedDRA- |
Häufigkeit der ADR* |
|
und des Unterhautzellgewebes |
Angioödeme, Dermatitis, Erytheme, flüchtige Hautrötungen |
unbekannt |
|
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen |
Rückenschmerzen, Muskelkrämpfe, Schmerzen in den Extremitäten |
gelegentlich |
|
Arthralgie, Myalgie |
unbekannt | |
|
Erkrankungen der Nieren und Harnwege |
Nierenversagen |
unbekannt |
|
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort |
Müdigkeit, Schüttelfrost, Fieber |
häufig |
|
Brustschmerzen, Krankheitsgefühl, Schmerzen, Brustbeschwerden, Unwohlsein, Kältegefühl, Hitzegefühl, grippeähnliche Erkrankungen, Rötungen an der Infusionsstelle, Extravasate an der Infusionsstelle, Schmerzen an der Infusionsstelle |
gelegentlich | |
|
Reaktionen an der Infusionsstelle, Gangstörungen, Ödeme |
unbekannt | |
|
Untersuchungen |
erhöhter Blutdruck |
gelegentlich |
|
positiver direkter Coombs-Test |
unbekannt | |
*Die Häufigkeit wurde anhand der folgenden Kriterien bestimmt: sehr häufig (> 1/10), häufig (>1/100, <1/10), gelegentlich (>1/1 000, <1/100), selten (>1/10 000, <1/1 000), sehr selten (< 1/10 000).
Pädiatrische Population:
Es wird davon ausgegangen, dass die bei Kindern auftretenden Nebenwirkungen hinsichtlich Häufigkeit, Art und Schweregrad den bei Erwachsenen beobachteten Nebenwirkungen entsprechen.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel, Paul-Ehrlich-Institut, Paul-EhrlichStraße 51 - 59, 63225 Langen, Telefon +49 6 10 37 70, Telefax: +49 61 03 77 12 34, Website: www.pei.de anzuzeigen.
4.9. Überdosierung
Überdosierung kann zur Volumenüberlastung und Hyperviskosität führen besonders bei Risikopatienten, einschließlich älteren Patienten und Patienten mit eingeschränkter Herz-oder Nierenfunktion.
Pädiatrische Population:
Bei Kindern und Jugendlichen mit Risikofaktoren, z.B. eingeschränkter Herz- oder Nierenfunktion, kann Überdosierung wie bei anderen intravenösen Immunglobulinen zu Flüssigkeitsüberlastung und Hyperviskosität führen.
5. PHARMAKOLOGISCHE EIGENSCHAFTEN
5.1. Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Sera und Immunglobuline/normales Immunglobulin vom Menschen zur intravenösen Anwendung.
ATC-Code: J06BA02
Gammagard S/D enthält hauptsächlich funktionell intaktes Immunglobulin G (IgG) mit einem breiten Antikörperspektrum gegen infektiöse Erreger.
Gammagard S/D enthält das komplette Immunglobulin G-Antikörperspektrum, das in der Normalbevölkerung vorhanden ist. Es wird aus gepooltem Plasma von mindestens 1000 Spenden hergestellt. Die Verteilung der Immunglobulin G-Subklassen entspricht nahezu der des natürlichen, menschlichen Plasmas. Durch Verabreichung entsprechender Dosen von Gammagard S/D werden abnormal verminderte IgG-Spiegel wieder in den Normalbereich angehoben.
Der Wirkmechanismus von Immunglobulinen bei anderen Indikationen als der Substitutionstherapie ist nicht vollständig aufgeklärt, schließt aber immunmodulatorische Effekte mit ein.
Pädiatrische Population:
Es ist davon auszugehen, dass die pharmakodynamischen Eigenschaften bei Kindern und Jugendlichen dieselben sind, wie bei Erwachsenen.
5.2. Pharmakokinetische Eigenschaften
Gammagard S/D ist nach intravenöser Applikation sofort und vollständig im Kreislauf des Patienten bioverfügbar. Es verteilt sich relativ schnell zwischen Plasma und extravaskulärer Flüssigkeit; das Gleichgewicht zwischen Intra-und Extravasalraum ist nach etwa 3-5 Tagen erreicht.
Die Halbwertszeit von Gammagard S/D beträgt etwa 37,7 ± 15 Tage. Sie kann von Patient zu Patient variieren, besonders bei primären Immundefekten.
IgG und IgG-Komplexe werden in den Zellen des retikuloendothelialen Systems abgebaut.
5.3. Präklinische Daten zur Sicherheit
Immunglobuline sind normale Bestandteile des menschlichen Körpers.
Die Sicherheit von Gammagard S/D wurde in mehreren nicht-klinischen Studien nachgewiesen.
Basierend auf den konventionellen Studien zur Sicherheitspharmakologie und Genotoxizität lassen die nicht-klinischen Daten keine besonderen Risiken für den Menschen erkennen.
Da keine klinischen Hinweise auf ein kanzerogenes Potenzial von Immunglobulinen vorliegen, sind keine experimentellen Studien mit heterogenen Spezies durchgeführt worden.
6. PHARMAZEUTISCHE ANGABEN
6.1. Liste der sonstigen Bestandteile
Pulver: Humanalbumin (0,6 g/g IgG)
Glyzin
Natriumchlorid
Glukosemonohydrat
Lösungsmittel: Sterilisiertes Wasser für Injektionszwecke
6.2. Inkompatibilitäten
Gammagard S/D darf nicht mit anderen Arzneimitteln vermischt werden.
6.3. Dauer der Haltbarkeit 2 Jahre
Die chemische und physikalische Stabilität der gebrauchsfertigen Lösung ist über 2 Stunden bei Raumtemperatur belegt. Aus mikrobiologischer Sicht sollte die Lösung dennoch unmittelbar verwendet werden, außer die Herstellung der gebrauchsfertigen Lösung schließt das Risiko einer mikrobiologischen Kontamination aus. Wird die gebrauchsfertige Lösung nicht unverzüglich verwendet, liegen Lagerbedingungen und -zeit in der Verantwortung des Anwenders und sollte normalerweise nicht länger als 24 Stunden bei 2-8 °C betragen, wenn die Rekonstitution unter kontrollierten und validierten, aseptischen Bedingungen stattgefunden hat.
6.4. Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Nicht über 25°C lagern.
Nicht Einfrieren, um ein Zerbrechen der Lösungsmitteldurchstechflasche zu vermeiden. Durchstechflaschen im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Nach Ablauf des Verfalldatums nicht mehr verwenden.
Für Kinder unzugänglich aufbewahren.
6.5. Art und Inhalt des Behältnisses
Alle Pulver und Lösungsmittel-Zubereitungen sind in Durchstechflaschen aus Glas Typ I abgefüllt. Die Pulver-und Lösungsmittel-Durchstechflaschen sind mit Gummistopfen aus Bromobutyl mit Silikonüberzug verschlossen.
Gammagard S/D ist in Packungsgrößen zu 0,5 g, 2,5 g, 5 g und 10 g erhältlich.
Jede Gammagard S/D Packung zu 0,5 g enthält das Lösungsmittel (10 ml), eine sterile doppelseitige Nadel (Transfernadel), eine sterile Filternadel, eine 10 ml-Plastikspritze und ein steriles MiniInfusionsset.
Jede Gammagard S/D Packung zu 2,5 g, 5 g und 10 g enthält das Lösungsmittel (50 ml, 96 ml, 192 ml), ein steriles Überleitungsgerät und ein steriles Infusionsbesteck mit Filter.
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
6.6. Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung
Bei aseptischer Zubereitung der Lösung außerhalb einer sterilen Werkbank sollte die Verabreichung so schnell wie möglich, auf jeden Fall aber innerhalb von 2 Stunden nach Rekonstitution erfolgen. Bei aseptischer Zubereitung in einer sterilen Werkbank kann die Lösung bei konstanter Kühlung (2 °C bis 8 °C) bis zu 24 Stunden aufbewahrt werden. Wenn diese Bedingungen nicht zutreffen, ist die Sterilität der rekonstituierten Lösung nicht gewährleistet. Angebrochene Durchstechflaschen sollten verworfen werden.
Nach Zugabe des Lösungsmittels sollte die Auflösung der Trockensubstanz innerhalb von 30 Minuten erfolgen. Die Lösung sollte vor Verabreichung auf Raum-oder Körpertemperatur angewärmt werden.
Vor der Rekonstitution sollte die Trockensubstanz ein weißes(r) oder ganz schwachgelbes(r) Pulver/Kuchen sein das (der) frei von sichtbaren Fremdkörpern ist. Das rekonstituierte Produkt sollte vor der Verabreichung visuell auf sichtbare Teilchen oder Farbveränderungen überprüft werden. Die Lösung sollte klar oder leicht opaleszierend, farblos oder schwachgelb sein. Trübe Lösungen oder solche mit Niederschlägen nicht verwenden.
Nicht verbrauchtes Arzneimittel oder Abfallmaterialien sollten in Übereinstimmung mit den lokalen Bestimmungen entsorgt werden.
Lösen des Arzneimittels - aseptische Zubereitung:
Gammagard S/D 0,5 g
Gammagard S/D und das Sterilisierte Wasser für Injektionszwecke (Lösungsmittel) auf Raumtemperatur erwärmen. Diese Temperatur sollte beibehalten werden, bis die Lösung des Arzneimittels abgeschlossen ist.
A) 5%ige Lösung
1. Die Kappen von den Fläschchen entfernen und die Gummistopfen mit einer keimtötenden Flüssigkeit reinigen.
2. Die Schutzhülle von einem Ende der Transfernadel entfernen. Freies Ende nicht berühren!
3. Die Lösungsmittelflasche auf eine flache Oberfläche stellen. Die Transfernadel senkrecht in die Mitte des Gummistopfens einstechen.
4. Schutzhülle vom anderen Ende der Transfernadel entfernen. Freies Ende nicht berühren!
5. Die Lösungsmittelflasche mit der aufgesetzten Transfernadel in einem bestimmten Winkel zu der Konzentratflasche halten, um ein Verschütten des Lösungsmittels zu vermeiden.
6. Die Konzentratflasche in der Mitte des Gummistopfens durchstechen, dabei die Lösungsmittelflasche rasch kopfüber umdrehen, ohne das Lösungsmittel zu verschütten. ACHTUNG: Wenn der Dorn nicht genau in die Mitte des Gummistopfens eingestochen wird, kann das ein Verrutschen des Stopfens und damit ein Verlust des Vakuums zur Folge haben.
7. Wenn der Transfer des Lösungsmittels abgeschlossen ist, die leere Lösungsmittelflasche mit der Transfernadel von der Trockensubstanzflasche abziehen. Die Trockensubstanzflasche vorsichtig schwenken, um den Inhalt sorgfältig zu mischen.
ACHTUNG: Nicht Schütteln. Schaumbildung vermeiden.
8. Transfernadel nach der einmaligen Verwendung verwerfen.
B) 10%ige Lösung
1. Die Kappen von den Fläschchen entfernen und die Gummistopfen mit einer keimtötenden Flüssigkeit reinigen.
2. Die Trockensubstanz mit der entsprechenden Menge Lösungsmittel mittels einer sterilen, hypodermischen Nadel und einer Spritze rekonstituieren. Das Lösungsmittelvolumen zur Herstellung einer 10%igen Lösung beträgt 5 ml bei der Packungsgröße 0,5 g.
3. Unter aseptischen Bedingungen das Lösungsmittel mit einer sterilen, hypodermischen Nadel in eine Spritze aufziehen. Das Lösungsmittel wird dann in die Trockensubstanzflasche übergeführt.
4. Die Trockensubstanzflasche sofort sorgfältig schwenken, um den Inhalt sorgfältig zu mischen ACHTUNG: Nicht Schütteln. Schaumbildung vermeiden.
5. Transfernadel nach der einmaligen Verwendung verwerfen.
Gammagard S/D 2,5 g, 5 g und 10 g
Gammagard S/D und das Sterilisierte Wasser für Injektionszwecke (Lösungsmittel) auf Raumtemperatur erwärmen. Diese Temperatur sollte beibehalten werden, bis die Lösung des Arzneimittels abgeschlossen ist.
A) 5%ige Lösung
1. Die Kappen von den Fläschchen entfernen und die Gummistopfen mit einer keimtötenden Flüssigkeit reinigen.
2. Die Schutzhülle von einem Ende der Transfernadel entfernen und die Nadel durch den Gummistopfen der Lösungsmittelflasche stechen. Freies Ende nicht berühren!
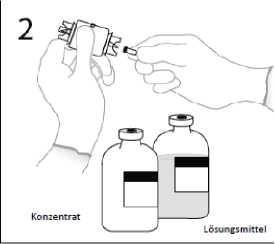
3a. Die Lösungsmittelflasche auf eine flache Oberfläche stellen. Die Transfernadel senkrecht in die Mitte des Gummistopfens einstechen.
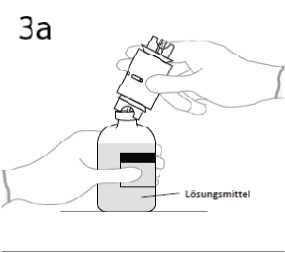
ACHTUNG: Wenn der Dorn nicht genau in die Mitte des Gummistopfens eingestochen wird, kann das ein Verrutschen des Stopfens und ein Verlust des Vakuums zur Folge haben.
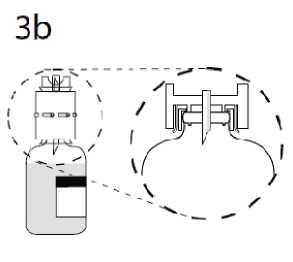
3b. Durch sorgfältiges Niederdrücken des Überleitungsgerätes sicherstellen, dass der Rand vollständig in das Medizinprodukt eindringt.
Während des Halten an dem Überleitungsgerät die Kappe des überstehenden Dorns entfernen. Freies Ende nicht berühren!
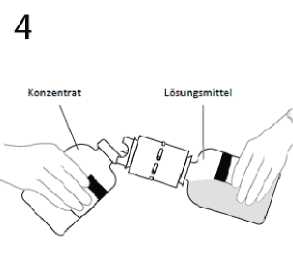
4. Lösungsmittelflasche mit dem Ende der aufgesetzten Transfemadel in einem bestimmten Winkel
zur Konzentratflasche halten, um ein Verschütten des Lösungsmittels zu vermeiden.
Hinweis: Die Lösungsmittelflasche nicht umdrehen, da dies zu einem Auslaufen der Lösung führen kann.
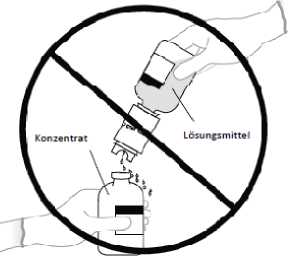
5 a. Die Konzentratflasche in der Mitte des Gummistopfens durchstechen, dabei die
Lösungsmittelflasche rasch kopfüber umdrehen, ohne das Lösungsmittel zu verschütten.
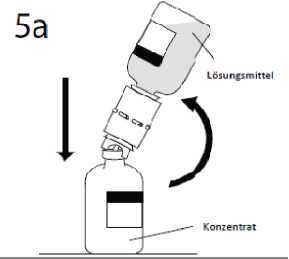
ACHTUNG: Wenn der Dorn nicht genau in die Mitte des Gummistopfens eingestochen wird, kann das ein Verrutschen des Stopfens und damit ein Verlust des Vakuums zur Folge haben.
5b. Durch sorgfältiges Niederdrücken des Überleitungsgerätes sicherstellen, dass der Rand vollständig in das Medizinprodukt eindringt.
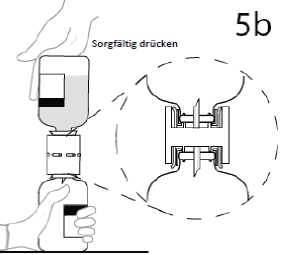
6. Wenn der Transfer des Lösungsmittels abgeschlossen ist, die leere Lösungsmittelflasche mit der Transfemadel von der Trockensubstanzflasche abziehen. Die Trockensubstanzflasche vorsichtig schwenken, um den Inhalt sorgfältig zu mischen.
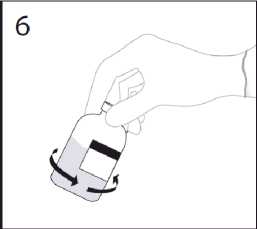
ACHTUNG: Nicht Schütteln. Schaumbildung vermeiden.
Transfernadel nach der einmaligen Verwendung verwerfen.
B) 10%ige Lösung
1. Schritt 1 wie unter Punkt A beschrieben durchführen.
2. Für die Herstellung der 10%igen Lösung die halbe Menge des Lösungsmittels verwenden. Tabelle 2 zeigt die Volumina, die aus der Lösungsmittelflasche entnommen werden sollten, bevor das Überleitungsgerät angeschlossen wird um die 10%ige Lösung herzustellen. Aseptische Bedingungen einhalten; das nicht-benötigte Lösungsmittelvolumen mit einer sterilen, hypodermischen Nadel und Spritze entnehmen. Die gefüllte Spritze verwerfen.
3. Mit dem verbleibenden Lösungsmittel im Lösungsmittelfläschchen verfahren Sie wie unter den Punkten 2-6 unter A beschrieben.
TABELLE 2
Nicht-benötigtes Lösungsmittel-Volumen
2,5 g 5 g 10 g
Konzentration
5%
10 %
Fläschchen Fläschchen Fläschchen
Kein Lösungsmittel zur Herstellung der 5%igen Lösung entfernen
25 ml 48 ml 96 ml
Bei Verabreichung auf aseptische Arbeitsweise achten!
Gammagard S/D 0,5 g
1. Filternadel auf eine sterile, leere Kunststoffspritze aufsetzen und festdrehen.
2. Den Spritzenkolben zurückziehen, damit Luft in die Spritze gelangen kann.
3. Das Gammagard S/D Fläschchen mit der gelösten Trockensubstanz auf eine ebene Fläche stellen und fest halten. Die Filternadel senkrecht durch die Mitte des Stopfens stechen.
4. Erst Luft in das Fläschchen injizieren und dann die Lösung in die Spritze aufziehen.
5. Die Spritze abnehmen und das Mini-Infusionsset an die gefüllte Spritze anschließen. Die fertige Lösung intravenös injizieren.
Gammagard S/D 2,5 g, 5 g und 10 g
In jeder Packung ist ein Infusionsbesteck mit Gebrauchsinformation. Bei Verwendung eines anderen Infusionsbestecks sicherstellen, dass das Set einen vergleichbaren Filter hat.
7. INHABER DER ZULASSUNG
Baxter Deutschland GmbH Edisonstraße 4 85716 Unterschleißheim Telefon: 089/31701-0 Fax: 089/31701-177 E-Mail: info_de@baxter.com Internet: www.baxter.de
8. ZULASSUNGSNUMMER
186a/92
9. DATUM DER ERTEILUNG DER ZULASSUNG/VERLÄNGERUNG DER ZULASSUNG
Datum der Zulassung: 14. April 1994
Datum der Verlängerung der Zulassung: 27. Januar 2005
10. STAND DER INFORMATION
April 2014
11. HERKUNFTSLÄNDER DER ZUR PRODUKTION VERWENDETEN PLASMEN
Deutschland, Finnland, Norwegen, Österreich, Schweden, Schweiz, Tschechien und Vereinigte Staaten von Amerika
12. VERKAUFSABGRENZUNG
Verschreibungspflichtig
Baxter und Gammagard S/D sind eingetragene Marken der Baxter International Inc., registriert unter US-Patent und dem US-Handelsgesetz.
17