Gamunex 10%
1. BEZEICHNUNG DES ARZNEIMITTELS
Gamunex 10% - Infusionslösung
2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
Wirkstoff: Normales Immunglobulin vom Menschen (IVIg)
Ein ml enthält:
normales Immunglobulin vom Menschen..........................................................100 mg
(IgG-Reingehalt von mindestens 98%)
Eine Durchstechflasche mit 10 ml enthält: 1 g Eine Durchstechflasche mit 50 ml enthält: 5 g Eine Durchstechflasche mit 100 ml enthält: 10 g Eine Durchstechflasche mit 200 ml enthält: 20 g
Anteile der IgG-Subklassen (Durchschnittswerte):
|
IgG1.......... |
..........62,8% |
|
IgG2.......... |
..........29,7% |
|
IgG3.......... |
............4,8% |
|
IgG4.......... |
.............2,7% |
1 ml Gamunex 10% enthält 100 mg Protein mit einem IgG-Anteil von mindestens 98%
(IgA-Gehalt: durchschnittlich: 59 Mikrogramm/ml; max.: 84 Mikrogramm/ml;
Analysenergebnisse von 5 unterschiedlichen Chargen).
Hergestellt aus humanem Spenderplasma.
Vollständige Auflistung der sonstigen Bestandteile siehe Abschnitt 6.1
3. DARREICHUNGSFORM
Infusionslösung.
4. KLINISCHE ANGABEN
4.1 Anwendungsgebiete
Substitutionstherapie bei Erwachsenen und Kindern und Jugendlichen (0-18 Jahre):
- Primäre Immunmangelsyndrome mit verminderter Antikörperproduktion (siehe Abschnitt 4.4).
- Hypogammaglobulinämie und rezidivierende bakterielle Infektionen bei Patienten mit chronisch-lymphatischer Leukämie, bei denen sich eine prophylaktische Antibiotikagabe als unwirksam erwiesen hat.
- Hypogammaglobulinämie und rezidivierende bakterielle Infektionen bei Patienten in der Plateauphase eines Multiplen Myeloms, die auf eine PneumokokkenImmunisierung nicht angesprochen haben.
- Hypogammaglobulinämie bei Patienten nach Transplantation allogener hämatopoetischer Stammzellen (allogene HSCT).
- Kongenitales AIDS mit rezidivierenden bakteriellen Infektionen.
Immunmodulation bei Erwachsenen sowie Kindern und Jugendlichen (0-18 Jahre) mit:
Primärer Immunthrombozytopenie (ITP) bei Patienten mit einem hohen Blutungsrisiko oder zur Korrektur der Thrombozytenzahl vor chirurgischen Eingriffen.
Guillain-Barre-Syndrom.
Kawasaki-Syndrom.
Chronisch Inflammatorischer Demyelinisierender Polyneuropathie (CIDP)
4.2 Dosierung und Art der Anwendung
Eine Substitutionstherapie sollte stets unter der Aufsicht eines in der Behandlung von Immunmangelsyndromen erfahrenen Arztes eingeleitet und überwacht werden.
Dosierung
Die Dosierung und das Verabreichungsschema richten sich nach dem Anwendungsgebiet.
In der Substitutionstherapie sollte die Dosierung in Abhängigkeit von vorliegenden pharmakokinetischen Parametern und der klinischen Reaktion individuell angepasst werden. Folgende Dosierungen werden empfohlen:
Substitutionstherapie bei primären Immunmangelsyndromen:
Das Dosierungsschema sollte einen IgG-Talspiegel (gemessen vor der nächsten Infusion) von mindestens 5-6 g/l gewährleisten. Der Zeitraum bis zum Erreichen eines Gleichgewichts beträgt 3-6 Monate nach Beginn der Behandlung. Die empfohlene Anfangsdosis beträgt einmal 0,4-0,8 g/kg, gefolgt von einer Dosis von mindestens 0,2 g/kg in 3- bis 4-wöchentlichen Abständen.
Die benötigte Dosis, mit der IgG-Talspiegel von 5-6 g/l erreicht werden, liegt bei 0,2-0,8 g/kg/Monat. Nach Erreichen von steady-state--Bedingungen beträgt das erforderliche Dosierungsintervall 3-4 Wochen. Talspiegel müssen unter Berücksichtigung der Infektionsinzidenz gemessen und interpretiert werden. Zur Senkung der Infektionsrate kann eine Dosissteigerung erforderlich sein, um höhere Talspiegel zu erzielen.
Hypogammaglobulinämie und rezidivierende bakterielle Infektionen bei Patienten mit chronisch-lymphatischer Leukämie, bei denen sich eine prophylaktische Antibiotikagabe als unwirksam erwiesen hat;
Hypogammaglobulinämie und rezidivierende bakterielle Infektionen bei Patienten in der Plateauphase eines Multiplen Myeloms, die nicht auf eine PneumokokkenImmunisierung angesprochen haben;
Kongenitales AIDS mit rezidivierenden bakteriellen Infektionen:
Die empfohlene Dosis beträgt 0,2-0,4 g/kg alle drei bis vier Wochen.
Hypogammaglobulinämie bei Patienten nach Transplantation allogener hämatopoetischer Stammzellen
Die empfohlene Dosis beträgt 0,2-0,4 g/kg alle drei bis vier Wochen. Die Talspiegel sollten oberhalb von 5 g/l gehalten werden.
Primäre Immunthrombozytopenie
Es stehen zwei alternative Behandlungsschemata zur Verfügung:
- Einmal 0,8-1 g/kg an Tag 1; diese Dosis kann innerhalb von 3 Tagen einmalig wiederholt werden.
- Einmal täglich 0,4 g/kg über zwei bis fünf Tage.
Die Behandlung kann im Fall eines Rezidivs wiederholt werden.
Guillain-Barre-Syndrom
0,4 g/kg/Tag über 5 Tage.
Chronisch Inflammatorische Demyelinisierende Polyneuropathie:
2 g/kg (20 ml/kg) in geteilten Dosen über 2 bis 4 aufeinanderfolgende Tage als Initialdosis. Als Erhaltungsdosis wird eine Verabreichung von 1 g/kg innerhalb eines Tages (10 ml/kg) oder zwei geteilte Dosen von 0,5 g/kg (5 ml/kg KG) an zwei aufeinander folgenden Tagen, alle 3 Wochen empfohlen.
Die Erfahrung bei der Behandlung von Kindern mit Chronischer Inflammatorischer Demyelinisierender Polyneuropathie mit intravenösen Immunglobulinen ist begrenzt. Für die Bestimmung eines genauen Behandlungsergebnisses bei Patienten > 65 Jahre wurden nicht genügend Patienten in die klinischen Studien mit Gamunex 10% eingeschlossen.
Kawasaki-Syndrom:
Es sollten entweder 1,6-2 g/kg verteilt auf mehrere Einzeldosen über zwei bis fünf Tage oder einmalig 2 g/kg als Einzeldosis verabreicht werden. Die Patienten sollten zusätzlich eine Therapie mit Acetylsalicylsäure erhalten.
Die Dosierungsempfehlungen werden in der folgenden Tabelle zusammengefasst:
|
Indikation |
Dosis |
Häufigkeit der Verabreichung |
|
Substitutionstherapie bei primären Immunmangelsyndromen |
- Anfangsdosis: 0,4-0,8 g/kg - danach: 0,2-0,8 g/kg |
alle 3-4 Wochen, um einen Serum-Talspiegel von mind. 5-6 g/l zu erreichen |
|
Substitutionstherapie bei sekundären Immunmangelkrankheiten |
0,2-0,4 g/kg |
alle 3-4 Wochen, um einen Serum-Talspiegel von mind. 5-6 g/l zu erreichen |
|
Kongenitales AIDS |
0,2-0,4 g/kg |
alle 3-4 Wochen |
|
Hypogammaglobulinämie (<4 g/l) bei Patienten nach Transplantation allogener hämatopoetischer Stammzellen |
0,2-0,4 g/kg |
alle 3-4 Wochen, um einen Serum-Talspiegel von mind. 5 g/l zu erreichen |
|
Immunmodulation: - Primäre Immunthrombozytopenie |
0,8-1 g/kg oder |
an Tag 1; kann innerhalb von 3 Tagen einmalig wiederholt werden |
|
0,4 g/kg/d |
an 2-5 Tagen | |
|
- Guillain-Barre-Syndrom |
0,4 g/kg/d |
über 5 Tage |
|
- Chronisch Inflammatorische Demyelinisierende Polyneuropathie #: |
Initialdosis 2 g/kg Erhaltungsdosis 1 g/kg |
in geteilten Dosen über 2 bis 4 aufeinander folgende Tage Verabreichung innerhalb eines Tages oder aufgeteilt in 2 Dosen von 0,5 g/kg (5 ml/kg), verabreicht an 2 aufeinander folgenden Tagen, alle 3 Wochen |
|
- Kawasaki-Syndrom |
1,6-2 g/kg oder |
verteilt auf mehrere Einzeldosen über 2 bis 5 Tage zusätzlich zur Acetylsalicylsäure-Therapie |
|
2 g/kg |
als Einzeldosis zusätzlich zur Acetylsalicylsäure-Therapie |
#
Die Dosis beruht auf der angewendeten Dosis in der klinischen Studie mit Gamunex 10%.
Eine Behandlungsdauer über 48 Wochen hinaus sollte im Ermessen des Arztes liegen, abhängig vom Ansprechen des Patienten und das Ansprechen in der Langzeitanwendung.
Im individuellen Krankheitsverlauf kann eine Anpassung der Dosis und des Dosisintervalls erforderlich sein.
Kinder und Jugendliche
Die Dosierung bei Kindern und Jugendlichen (0-18 Jahre) unterscheidet sich nicht von derjenigen bei Erwachsenen, da die Dosierungsangaben für alle Anwendungsgebiete sich auf das Körpergewicht beziehen und die Dosierung anhand des klinischen Verlaufs der o.g. Erkrankungen angepasst wird.
Art der Anwendung
Zur intravenösen Anwendung.
Normales Immunglobulin vom Menschen sollte initial über einen Zeitraum von einer halben Stunde mit einer Infusionsgeschwindigkeit von 0,6-1,2 ml/kg/h intravenös verabreicht werden. Bei guter Verträglichkeit (siehe Abschnitt 4.4) kann die Infusionsgeschwindigkeit schrittweise bis auf maximal 4,8-8,4 ml/kg/h erhöht werden.
4.3 Gegenanzeigen
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile. Dies gilt insbesondere für Patienten mit anti-IgA-Antikörpern.
4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Bei Anwendung hoher Infusionsraten (8,4 ml/kg/h) müssen alle Patienten engmaschig überwacht werden. Bei Kindern sowie bei Patienten mit einem erhöhten Risiko für eine akute Niereninsuffizienz sollte die Infusionsgeschwindigkeit einen Maximalwert von 4,8 ml/kg/h nicht überschreiten.
Gamunex 10% darf nicht mit anderen Infusionslösungen (z. B. Kochsalzlösung) oder anderen Arzneimitteln gemischt werden. Sollte eine Verdünnung erforderlich sein, kann hierfür 5%-ige (50 mg/ml) Glucoselösung verwendet werden. In Fällen von latentem Diabetes (wenn vorübergehende Glykosurie auftreten kann), bei Diabetes oder bei Patienten, die eine zuckerarme Diät einhalten müssen, sollte die Anwendung von 50 mg/ml Glucoselösung sorgfältig überwacht werden. Siehe auch unten Warnhinweis zu akuter Niereninsuffizienz.
Gleichzeitige Verabreichung von Gamunex 10% und Heparin über dieselbe Infusionsleitung muss vermieden werden.
Bestimmte schwere Nebenwirkungen können in Zusammenhang mit der Infusionsgeschwindigkeit stehen. Die in Abschnitt 4.2 angegebene empfohlene Infusionsgeschwindigkeit muss strikt eingehalten werden. Die Patienten müssen während der Infusion engmaschig überwacht und im Hinblick auf eventuell auftretende Symptome sorgfältig beobachtet werden.
Bestimmte Nebenwirkungen können häufiger auftreten bei:
- einer hohen Infusionsgeschwindigkeit,
- Patienten, die erstmals normales Immunglobulin G vom Menschen erhalten, oder, in seltenen Fällen, bei einem Wechsel des normalen HumanimmunglobulinPräparates oder wenn die letzte Infusion längere Zeit zurückliegt.
Mögliche Komplikationen können häufig vermieden werden, wenn sichergestellt wird, dass die Patienten:
- keine Überempfindlichkeit gegenüber normalem Immunglobulin vom Menschen aufweisen, indem ihnen das Präparat anfangs langsam infundiert wird (0,1 ml/kg/h)),
- während der gesamten Dauer der Infusionszeit sorgfältig auf Symptome hin überwacht werden. Insbesondere solche Patienten, die erstmals menschliches Immunglobulin erhalten, die von einem anderen Immunglobulin-Präparat umgestellt werden oder bei denen seit der letzten Infusion ein längerer Zeitraum vergangen ist, sollten während der ersten Infusion sowie während der ersten Stunde nach der ersten Infusion überwacht werden, um mögliche Nebenwirkungen festzustellen. Alle anderen Patienten sollten im Anschluss an die Verabreichung mindestens 20 Minuten lang überwacht werden.
Bei Eintreten von Nebenwirkungen muss entweder die Infusionsgeschwindigkeit reduziert oder die Infusion gestoppt werden. Die erforderliche Therapie richtet sich nach der Art und dem Schweregrad der jeweiligen Nebenwirkung. Bei einem Schock ist eine Schockbehandlung gemäß den geltenden klinischen Standards einzuleiten.
Die intravenöse Verabreichung von Immunglobulinen erfordert bei allen Patienten:
• Eine adäquate Auffüllung des Flüssigkeitshaushaltes vor Beginn der Immunglobulin-Infusion
• Eine Überwachung der Urinausscheidung
• Eine Überwachung des Kreatininspiegels im Serum
• Die Vermeidung einer gleichzeitigen Anwendung von Schleifendiuretika.
Dieses Arzneimittel ist nahezu „natriumfrei“.
Überempfindlichkeit
Echte Überempfindlichkeitsreaktionen sind selten. Sie können bei Patienten mit Anti-IgA-Antikörpern auftreten.
Die intravenöse Verabreichung von Immunglobulinen ist bei Patienten mit einem isolierten IgA-Mangel nicht indiziert, sofern der IgA-Mangel die einzige klinisch relevante Anomalie darstellt.
In seltenen Fällen können normale Immunglobuline vom Menschen einen Blutdruckabfall mit anaphylaktischen Reaktionen verursachen, auch wenn der Patient eine frühere Behandlung mit normalem Immunglobulin vom Menschen vertragen hat.
Thromboembolie
Es liegen klinische Hinweise auf einen Zusammenhang zwischen der Verabreichung von intravenösem Immunglobulin und thromboembolischen Ereignissen wie Herzinfarkt, zerebralen Durchblutungsstörungen (einschließlich Schlaganfall), Lungenembolie und tiefer Venenthrombose vor. Diese sind wahrscheinlich auf einen relativen Anstieg der Blutviskosität während des hohen Einstroms von Immunglobulin bei Risikopatienten zurückzuführen. Bei der Verschreibung und Infusion von Immunglobulinen ist bei adipösen und Patienten mit bereits vorliegenden Risikofaktoren für thrombotische Ereignisse, wie fortgeschrittenes Alter, Bluthochdruck, Diabetes mellitus und einer Anamnese mit vaskulären Erkrankungen oder thrombotischen Ereignissen, Patienten mit angeborener oder erworbener Thrombophilie, Patienten mit längeren Phasen einer Immobilität, bei Patienten mit schwerer Hypovolämie und bei Patienten mit Krankheiten, welche die Blutviskosität erhöhen, besondere Vorsicht angezeigt.
Bei Patienten mit einem erhöhten Risiko für thromboembolische Ereignisse sollten intravenöse Immunglobuline mit der niedrigsten Infusionsrate und der niedrigsten noch möglichen Dosis verabreicht werden.
Akute Niereninsuffizienz
Bei Patienten, die eine intravenöse Therapie mit Immunglobulinen erhielten, wurde über Fälle von akutem Nierenversagen berichtet. In den meisten Fällen konnten Risikofaktoren wie eine vorbestehende Niereninsuffizienz, Diabetes mellitus, Hypovolämie, Übergewicht, gleichzeitige Behandlung mit nephrotoxischen Arzneimitteln oder ein Lebensalter von über 65 Jahren nachgewiesen werden.
Bei Auftreten einer Nierenfunktionsstörung sollte das Absetzen der Behandlung mit intravenösen Immunglobulinen erwogen werden.
Die Berichte über Nierenfunktionsstörungen und akutes Nierenversagen wurden mit der Anwendung vieler der zugelassenen Immunglobulinpräparate, die verschiedene sonstige Bestandteile wie Saccharose, Glucose und Maltose enthalten, in Verbindung gebracht; der Anteil der Präparate, die Saccharose als Stabilisator enthielten, war in diesen Fällen aber überproportional groß. Daher sollten Patienten mit einem erhöhten Risiko nur intravenöse Immunglobulinpräparate ohne diese sonstigen Bestandteile erhalten. Gamunex 10% enthält weder Saccharose noch Maltose oder Glucose.
Bei Patienten mit einem erhöhten Risiko für ein akutes Nierenversagen sollten intravenöse Immunglobuline mit der niedrigsten Infusionsrate und der niedrigsten noch möglichen Dosis verabreicht werden.
Aseptische Meningitis (AMS)
Im Zusammenhang mit der intravenösen Verabreichung von Immunglobulinen wurde über das Auftreten einer aseptischen Meningitis berichtet. Das Absetzen der intravenösen Immunglobulintherapie führte innerhalb von wenigen Tagen zu einer folgenlosen Rückbildung der aseptischen Meningitis. Dieses Syndrom beginnt für gewöhnlich innerhalb von wenigen Stunden bis zu 2 Tagen nach der intravenösen Therapie mit Immunglobulinen. Im Liquor finden sich häufig positive Befunde mit einer Pleozytose von mehreren Tausend Zellen pro mm3, wobei Zellen aus der granulozytären Reihe überwiegen, sowie erhöhte Proteinspiegel bis zu einigen hundert mg/dl. Eine aseptische Meningitis kann häufiger bei einer hoch dosierten intravenösen Therapie mit Immunglobulinen (2 g/kg) auftreten.
Hämolytische Anämie
Intravenöse Immunglobulinpräparate können Blutgruppen-Antikörper enthalten, die als Hämolysine wirksam werden und in vivo eine Beschichtung der roten Blutkörperchen mit Immunglobulinen bewirken können, die ihrerseits zu einer positiven direkten Antiglobulinreaktion (Coombs-Test) und in seltenen Fällen auch zu einer Hämolyse führen kann. Im Gefolge einer intravenösen Therapie mit
Immunglobulinen kann sich somit durch das verstärkte Entfernen von roten Blutkörperchen aus der Zirkulation eine hämolytische Anämie entwickeln. Patienten, die intravenöse Immunglobuline erhalten, müssen daher auf klinische Anzeichen und Symptome einer Hämolyse hin überwacht werden (siehe Abschnitt 4.8).
Die Entwicklung einer Hämolyse ist mit folgenden Risikofaktoren assoziiert: Hohe Dosen, verabreicht als Einzeldosis oder in Teildosen über mehrere Tage verteilt; andere Blutgruppe als Gruppe 0; zugrunde liegende Entzündung. Bei Patienten mit einer anderen Blutgruppe als Gruppe 0, die hohe Dosen aufgrund anderer Indikationen als PID erhalten, wird erhöhte Aufmerksamkeit empfohlen.
Bei Patienten, die eine Substitutionstherapie wegen PID erhielten, wurde nur selten eine Hämolyse beobachtet.
Einzelfälle von Hämolyse-assoziierter Niereninsuffizienz/Nierenversagen mit tödlichem Ausgang sind aufgetreten.
Wechselwirkungen mit serologischen Untersuchungen
Nach Infusion von Immunglobulinen kann es durch den vorübergehenden Anstieg der verschiedenen, passiv übertragenden Antikörper im Blut des Patienten zu falsch positiven Testergebnissen bei serologischen Untersuchungen kommen. Die passive Übertragung von Antikörpern gegen Erythrozytenantigene, z. B. A, B, D kann einige serologische Untersuchungen auf Erythrozyten-Antikörper (z. B. direkter Coombs-Test) beeinflussen.
Übertragbare Erreger
Standardmaßnahmen zur Verhütung von Infektionen durch die Verabreichung von Medikamenten, die aus menschlichem Blut oder Plasma hergestellt wurden, beinhalten Spenderauswahl, Testung einzelner Spenden und Plasmapools auf spezifische Infektionsmarker und Einführung effektiver Herstellungsschritte zur Inaktivierung/Eliminierung von Viren. Dennoch kann die Möglichkeit der Übertragung von Erregern bei der Verabreichung von Medikamenten, die aus menschlichem Blut oder Plasma hergestellt worden sind, nicht völlig ausgeschlossen werden. Dies trifft auch für bisher unbekannte oder neu auftretende Viren oder Erreger zu.
Die ergriffenen Maßnahmen werden als wirksam gegenüber umhüllten Viren wie humanes Immundefizienzvirus (HIV), Hepatitis B Virus (HBV) und Hepatitis C Virus (HCV) angesehen. Die Viruseliminierung/ -inaktivierung ist möglicherweise bei nicht umhüllten Viren wie Hepatitis A Virus (HAV) und Parvovirus B19 von begrenztem Wert.
Die klinische Erfahrung hat bestätigt, dass Hepatitis A- Viren oder Parvoviren B19 nicht durch Immunglobuline übertragen werden, weiterhin wird angenommen, dass der Gehalt an Antikörpern einen wichtigen Beitrag zur Virussicherheit leistet.
Die Verabreichung von Gamunex 10% und die Chargennummer sind in der Krankengeschichte zu dokumentieren, um den Zusammenhang zwischen dem Patienten und der Charge des Produktes zu gewährleisten.
Kinder und Jugendliche
Obwohl nur begrenzte Daten vorliegen, ist zu erwarten, dass für Kinder und Jugendliche die gleichen Warnhinweise, Vorsichtsmaßnahmen und Risikofaktoren gelten. Post-Marketing Analysen zeigen, dass IVIg-Hochdosisindikationen bei Kindern, insbesondere Kawasaki-Syndrom, mit einer erhöhten Rate hämolytischer Reaktionen im Vergleich zu anderen IVIg-Indikationen bei Kindern verbunden sind.
Ärzte sollten unbedingt eine Überprüfung der Hämoglobinwerte 24 bis 48 Stunden nach Abschluss der IVIg-Behandlung erwägen, falls Verdacht auf Hämolyse besteht. Ist eine erneute Behandlung notwendig, so wird dringend empfohlen, bei Verdacht auf Hämolyse die Hämoglobinwerte eine Woche nach IVIg-Gabe zu kontrollieren. Familien sollten aufgefordert werden, zurück in die Klinik zu kommen, sollte ihr Kind Anzeichen von Hämolyse, wie Blässe, Lethargie, dunkler Urin, Dyspnoe oder Palpitationen entwickeln.
4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Abgeschwächte Lebendimpfstoffe
Die Verabreichung von Immunglobulinen kann über einen Zeitraum von mindestens 6 Wochen bis zu 3 Monaten hinweg die Wirksamkeit von Lebendimpfstoffen, z. B. gegen Masern, Röteln, Mumps oder Windpocken, abschwächen. Nach Verabreichung dieses Präparates sollten vor einer Impfung mit einem abgeschwächten Lebendimpfstoff 3 Monate vergehen. Bei Impfstoffen gegen Masern kann dieser Effekt bis zu einem Jahr anhalten. Daher sollte bei Patienten, die gegen Masern geimpft werden, der Antikörperstatus getestet werden.
Kinder und Jugendliche
Obwohl keine speziellen Studien zur Erfassung von Wechselwirkungen bei Kindern und Jugendlichen durchgeführt wurden, sind keine Unterschiede zwischen Erwachsenen und Kindern zu erwarten.
4.6 Fertilität, Schwangerschaft und Stillzeit
Schwangerschaft
Über die Sicherheit dieses Arzneimittels bei Anwendung in der Schwangerschaft liegen keine kontrollierten Studien vor. Daher sollte es Schwangeren und stillenden Müttern nur mit Vorsicht gegeben werden. Intravenöse Immunglobuline passieren, insbesondere während des letzten Schwangerschaftsdrittels, nachweislich die Placenta. Die klinische Erfahrung mit Immunglobulinen lässt jedoch keine schädlichen Auswirkungen auf den Verlauf der Schwangerschaft oder den Fötus und das Neugeborene erwarten.
Stillzeit
Immunglobuline gehen in die Muttermilch über und können zum Schutz des Neugeborenen gegenüber Pathogenen, die über die Schleimhäute eindringen, beitragen.
Fertilität
Die klinischen Erfahrungen mit Immunglobulinen deuten nicht auf schädliche Auswirkungen auf die Fertilität hin.
4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen können durch bestimmte, im Zusammenhang mit Gamunex 10% auftretende Nebenwirkungen beeinträchtigt werden. Patienten, bei denen im Verlauf der Behandlung solche Nebenwirkungen auftreten, sollten nicht Auto fahren und keine Maschinen bedienen, bevor diese wieder abgeklungen sind.
4.8 Nebenwirkungen
Zusammenfassung des Sicherheitsprofils
Nebenwirkungen wie Schüttelfrost, Kopfschmerz, Schwindel, Fieber, Erbrechen, allergische Reaktionen, Übelkeit, Arthralgien, niedriger Blutdruck und moderate Lumbalgien können gelegentlich auftreten.
Selten können normale Immunglobuline vom Menschen einen plötzlichen Blutdruckabfall, in Einzelfällen auch einen anaphylaktischen Schock verursachen, auch wenn der Patient keine Überempfindlichkeit bei früheren Anwendungen gezeigt hat.
Unter normalen Immunglobulinen vom Menschen wurden Fälle einer reversiblen aseptischen Meningitis sowie seltene Fälle vorübergehender Hautreaktionen beobachtet. Insbesondere bei Patienten mit den Blutgruppen A, B und AB wurde über reversible hämolytische Reaktionen berichtet. In seltenen Fällen kann es nach hoch dosierter intravenöser Verabreichung von Immunglobulinen zu einer hämolytischen Anämie kommen, die eine Bluttransfusion erforderlich macht (siehe hierzu auch Abschnitt 4.4).
Ein Anstieg des Kreatininspiegels im Serum und/oder ein akutes Nierenversagen wurden beobachtet.
Sehr selten kann es zu thromboembolischen Reaktionen wie Myokardinfarkt, Schlaganfall, Lungenembolie und tiefer Venenthrombose kommen.
Zu den Sicherheitsaspekten bezüglich übertragbarer Erreger siehe Abschnitt 4.4.
Tabellarische Auflistung möglicher Nebenwirkungen
In der nachfolgenden Tabelle werden die Nebenwirkungen gemäß MedDRA-Systemorganklasse eingeordnet. Bei den Häufigkeitsangaben werden folgende Kategorien zugrunde gelegt:
Sehr häufig (> 1/10)
Häufig (> 1/100 bis < 1/10)
Gelegentlich (> 1/1 000 bis < 1/100)
Selten (> 1/10 000 bis < 1/1 000)
Sehr selten (< 1/10 000)
Nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar)
Innerhalb jeder Häufigkeitsgruppe werden die Nebenwirkungen nach abnehmendem Schweregrad angegeben
Seltene Unerwünschte Arzneimittelwirkungen (UAW) aus klinischen Studien zu
Gamunex 10%:
Hämolytische Anämie, Atemnot, Sinusitis, Hautexfoliation, Angstzustände, Muskelschmerzen, Anämie, Verdauungsstörungen, Quetschung, Dermatitis, Hitzewallungen, Steifheit des Bewegungsapparats, Palmarerythem, Aphonie.
Häufigkeit unerwünschter Arzneimittelwirkungen (UAW) in klinischen Studien zu Gamunex 10%:
|
MedDRA-Systemorganklasse |
Bevorzugter Begriff gemäß MedDRA |
Häufigkeitskategorie der UAW |
|
Untersuchungen |
Leukozytenzahl vermindert |
gelegentlich |
|
Erkrankungen des Nervensystems |
Kopfschmerzen |
häufig |
|
Schwindel |
gelegentlich | |
|
Erkrankungen der Haut und des Unterhautgewebes |
Urtikaria, Dermatitis, Juckreiz, Hautausschlag, |
gelegentlich |
|
Erkrankungen des Gastrointestinaltrakts |
Bauchschmerzen, Durchfall, Übelkeit, Erbrechen |
gelegentlich |
|
Gefäßerkrankungen |
Hypertonus, Hypotonus |
gelegentlich |
|
Erkrankungen der Atemwege, des Brustraums und des Mediastinums |
Pharyngitis, Husten, verstopfte Nase, Giemen (keuchende Atmung) |
gelegentlich |
|
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen |
Arthralgie, Rückenschmerzen, Nackenschmerzen, Schulterschmerzen |
gelegentlich |
|
Herzerkrankungen |
Brustschmerzen |
gelegentlich |
|
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort |
Fieber |
häufig |
|
Influenzaartige Erkrankungen Unwohlsein, Müdigkeit, Schüttelfrost, Abgeschlagenheit, Reaktionen am Injektionsort |
gelegentlich |
Kinder und Jugendliche
Bei Kindern und Jugendlichen sind keine Unterschiede im Auftreten unerwünschter Arzneimittelwirkungen zu erwarten.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-RisikoVerhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel, Paul-Ehrlich-Institut, Paul-Ehrlich-
Straße 51 - 59, 63225 Langen, Telefon: +49 6103 77 0, Telefax: +49 6103 77 1234, Website: www.pei.de anzuzeigen.
4.9 Überdosierung
Überdosierung kann zu einer Flüssigkeitsüberladung und zu Hyperviskosität führen; dies gilt insbesondere bei Risikopatienten einschließlich älterer Patienten und Patienten mit Herz- oder Niereninsuffizienz.
5. PHARMAKOLOGISCHE EIGENSCHAFTEN
5.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Immunsera und Immunglobuline; Immunglobuline, normal human, zur intravasalen Anwendung. ATC-Code: J06BA02 Normales Immunglobulin vom Menschen enthält nicht modifiziertes Immunglobulin G (IgG) mit einem breiten Spektrum an Antikörpern gegen verschiedene Infektionserreger.
Normales Immunglobulin vom Menschen enthält die in der Normalbevölkerung vorhandenen IgG-Antikörper. Es wird aus dem Plasma von mindestens 1.000 Blutspenden gewonnen. Der darin enthaltene prozentuale Anteil der einzelnen Immunglobulin-G-Subklassen entspricht annähernd den Werten des nativen menschlichen Plasmas. Mit adäquaten Dosen dieses Arzneimittels kann ein abnorm erniedrigter Immunglobulin-G-Spiegel wieder auf den normalen Wert angehoben werden. Für Anwendungsgebiete außerhalb der Substitutionstherapie wurde der Wirkmechanismus noch nicht vollständig geklärt, es wurden jedoch immunmodulierende Effekte nachgewiesen.
Klinische Studien mit Gamunex 10% an Patienten mit Chronisch Inflammatorischer Demyelinisierender Polyneuropathie (CIDP):
Im Rahmen der IVIG-C CIDP Wirksamkeitsstudie (ICE-Studie), einer doppelblinden, randomisierten, Placebo-kontrollierten Studie wurde die Wirksamkeit und Unbedenklichkeit von Gamunex 10% in der Behandlung von CIDP untersucht. Insgesamt wurden 117 randomisierte CIDP Patienten alle 3 Wochen, entweder mit Gamunex 10% oder Placebo behandelt. Als Initialdosis wurden 2 g/kg KG, als Erhaltungsdosis 1 g/kg KG verabreicht.
Responder-Raten (entsprechend einer Verbesserung gemäß INCAT Disability Score und der Aufrechterhaltung von >1 Verbesserung über die 24-wöchentliche Wirksamkeitsperiode) waren in der Gamunex 10% Gruppe signifikant höher (54%), im Vergleich zu der Placebogruppe (21%, p=0.0002). Die Muskelkraft gemäß MRC Score und Griffstärke, sowie das Empfindungsvermögen gemessen nach ISS Score verbesserten sich in der Gamunex 10%-Gruppe maßgeblich im Vergleich zu Placebo.
Im Hinblick auf den begrenzten Einschluss von Patienten > 65 Jahren in die Studie, konnte im INCAT Score keine genaue Behandlungswirkung gezeigt werden; hinsichtlich der Griffstärke, konnte eine statistisch signifikante Behandlungswirkung mit Gamunex 10% nachgewiesen werden.
Von den Respondern sprachen weniger als die Hälfte nach der Initialdosis (bis zur 3. Woche) auf die Behandlung an, wobei die meisten nach der zweiten Dosis (bis zur 6. Woche) ansprachen. Non-Responder erhielten nach Cross-over die Behandlung des anderen Behandlungsarms bis zu 24 Wochen.
Alle Responder wurden in einer Verlängerungsphase von weiteren 6 Monaten re-randomisiert und einer Erhaltungstherapie mit entweder Gamunex 10% oder Placebo unterzogen. In der Gruppe der ehemaligen Gamunex 10%-Responder zeigten die zu einer Placebo-Behandlung randomisierten Patienten eine signifikant höhere Rückfallrate (42%) gegenüber denen zu einer Behandlung mit Gamunex 10% randomisierten (13%, p=0.012).
Mit der ICE-Studie konnte die Kurzzeit- und Langzeitwirksamkeit von Gamunex 10% bei der Behandlung von CIDP nachgewiesen werden. Die Ergebnisse dieser Studie werden in der folgenden Tabelle zusammenfassend dargestellt.
|
Primärer Endpunkt und weitere Ergebnisse der I |
CE-Studie | ||
|
Gamunex 10% |
Placebo |
p | |
|
Responder-Rate während der Wirksamkeitsphase (primärer Endpunkt) |
54% |
21% |
0,0002 |
|
Rückfallwahrscheinlichkeit in der Verlängerungsphase |
13% |
45% |
0,013 |
|
Griffstärke (kPA)1 (Veränderung gegenüber Baseline) | |||
|
Dominante Hand |
13.2 |
1.5 |
0,0008 |
|
Nicht-dominante Hand |
13.3 |
4.3 |
0,005 |
|
Muskelkraft (MRC3 Summen-Score)1 (Veränderung gegenüber Baseline) |
3.3 |
0.2 |
0,001 |
|
Empfindungsvermögen (ISS4 Score)2 (Veränderung gegenüber Baseline) |
-1.2 |
0.2 |
0,021 |
1 Verbesserung wird durch einen positiven Wert indiziert
2 Verbesserung wird durch einen negativen Wert indiziert 3MRC: Medical Research Council
4ISS: INCAT Sensory Sum Score
Das Fertigprodukt ist auf einen schwach sauren pH-Wert eingestellt. Da Gamunex 10% eine niedrige Pufferkapazität besitzt, wird es während der Infusion schnell vom Blut neutralisiert. Auch nach Verabreichung hoher Dosen Gamunex 10% wurde keine Veränderung des Blut-pH-Wertes gemessen. Die Osmolalität beträgt 258 mOsmol/kg Lösung und liegt damit nahe am physiologischen Wert (285-295 mOsmol/kg).
5.2 Pharmakokinetische Eigenschaften
Normales Immunglobulin vom Menschen ist nach intravenöser Verabreichung in der Blutbahn des Empfängers unmittelbar und vollständig bioverfügbar. Es verteilt sich relativ rasch zwischen Plasma und extravaskulärer Flüssigkeit; nach circa 3 - 5 Tagen wird ein Gleichgewicht zwischen intra- und extravaskulärem Kompartiment erreicht.
Normales Immunglobulin vom Menschen hat bei Patienten mit primärem Antikörpermangelsyndrom eine Halbwertszeit von ca. 35 Tagen, die damit über dem in der Literatur beschriebenen Wert von 21 Tagen bei Gesunden liegt. Diese Halbwertszeit kann jedoch, insbesondere bei primärer Immundefizienz, von Patient zu Patient variieren.
IgG und IgG-Komplexe werden in Zellen des retikulo-endothelialen Systems abgebaut.
Kinder und Jugendliche
Bei Kindern und Jugendlichen sind keine Unterschiede in den pharmakokinetischen Eigenschaften zu erwarten.
5.3 Präklinische Daten zur Sicherheit
Immunglobuline sind normale Bestandteile des menschlichen Körpers. Da die Verabreichung von Immunglobulinen in Tierstudien zur Bildung von Antikörpern führen kann, liegen nur begrenzt präklinische Daten zur Sicherheit vor. In den durchgeführten akuten und subakuten Toxizitätstierstudien zeigte Gamunex 10% kein besonderes Risiko für Menschen.
6. PHARMAZEUTISCHE ANGABEN
6.1 Liste der sonstigen Bestandteile
Glycin.
6.2 Inkompatibilitäten
Dieses Arzneimittel darf nicht mit anderen als den in Abschnitt 6.6 genannten gemischt werden.
6.3 Dauer der Haltbarkeit
3 Jahre
6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Bei 2 °C - 8 °C (im Kühlschrank) lagern. Nicht einfrieren.
Das Produkt kann im Umkarton für einen einmaligen Zeitraum von bis zu 6 Monaten bei Zimmertemperatur aufbewahrt werden (nicht über 25 °C). In diesem Fall verfällt das Produkt am Ende der 6-Monatsfrist; das neue Verfallsdatum muss auf dem Umkarton und auf dem Flaschenetikett vermerkt werden. Das neue Verfallsdatum darf das aufgedruckte Verfallsdatum nicht überschreiten. Danach verbrauchen oder vernichten. Eine erneute Kühlung ist nicht möglich.
6.5 Art und Inhalt des Behältnisses
Lösung zur intravenösen Infusion:
Durchstechflaschen aus Typ I oder II-Glas mit Halobutyl-Isopren-Gummistopfen oder Chlorbutyl-Stopfen.
Packungsgrößen: 10 ml, 50 ml, 100 ml, 200 ml; Klinikpackungen.
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung
Das Produkt vor Gebrauch auf Raum- oder Körpertemperatur bringen. Die Lösung sollte klar oder leicht opaleszierend und farblos oder leicht gelblich gefärbt sein. Lösungen, die trüb sind oder einen Bodensatz aufweisen, dürfen nicht verwendet werden. Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu entsorgen. Nach Anbruch umgehend infundieren. Eine weitere Aufbewahrung ist auch im Kühlschrank wegen einer möglichen Besiedlung mit Bakterien nicht erlaubt.
Falls eine Verdünnung der Infusionslösung notwendig ist, kann hierzu eine 5%-ige Glucoselösung verwendet werden. Nicht mit Kochsalz-Lösung verdünnen. Gleichzeitige Gabe von Gamunex 10% mit Heparin über dieselbe Infusionsleitung muss vermieden werden.
Gamunex 10% Infusionsbestecke können mit 50 mg/ml Glucoselösung oder mit physiologischer Kochsalzlösung gespült werden. Sie dürfen nicht mit Heparin gespült werden.
Ein Heparin Lock, durch das Gamunex 10% verabreicht wurde, sollte mit 50 mg/ml Glucoselösung oder mit physiologischer Kochsalzlösung gespült werden. Es darf nicht mit Heparin gespült werden.
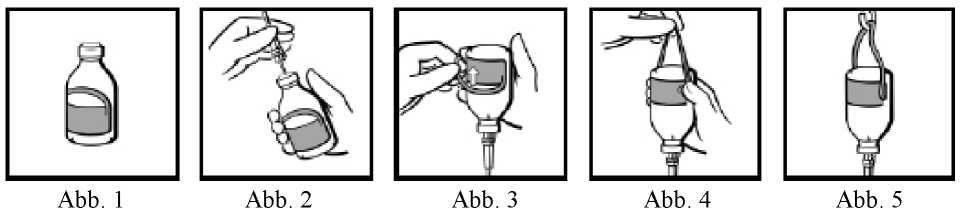
Hinweis zur Handhabung der Durchstechflaschen (nur 50 ml, 100 ml und 200 ml Flaschen)
Die Durchstechflaschen sind mit einem Hängeetikett versehen (Abb. 1). Nach Einführen des Infusionsbestecks (Abb. 2) drehen Sie bitte die Flasche um und ziehen Sie den Schlaufenteil des Etikettes ab (Abb. 3). Erzeugen Sie mit kräftigem Fingerdruck einen Knick an beiden Übergängen des Schlaufenteiles zum restlichen Etikett (Abb. 4). Hängen Sie die Durchstechflasche mit Hilfe der entstandenen Schlaufe am Infusionsständer auf (Abb. 5).
7.
INHABER DER ZULASSUNG
Grifols Deutschland GmbH Lyoner Straße 15 60528 Frankfurt Deutschland
Tel.: 069-660 593 100
8. ZULASSUNGSNUMMER PEI.H.02726.01.1
9. DATUM DER ERTEILUNG DER ZULASSUNG/VERLÄNGERUNG DER ZULASSUNG
09. Februar 2004
Datum der letzten Verlängerung der Zulassung: 08. Juni 2011
10. STAND DER INFORMATION
April 2014
11. ABGABESTATUS
Verschreibungspflichtig
12. HERKUNFTSLAND DES BLUTPLASMAS
USA