Goltor 10 Mg/10 Mg Tabletten
Fachinformation (Zusammenfassung der Merkmale der Arzneimittel)
1. BEZEICHNUNG DER ARZNEIMITTEL
GOLTOR® 10 mg/10 mg, 10 mg/20 mg, 10 mg/40 mg oder 10 mg/80 mg Tabletten
2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
Jede Tablette enthält 10 mg Ezetimib und 10 mg, 20 mg, 40 mg oder 80 mg Simvastatin.
Sonstige Bestandteile mit bekannter Wirkung:
Jede 10 mg/10 mg-Tablette enthält 58,2 mg Lactose-Monohydrat.
Jede 10 mg/20 mg-Tablette enthält 126,5 mg Lactose-Monohydrat.
Jede 10 mg/40 mg-Tablette enthält 262,9 mg Lactose-Monohydrat.
Jede 10 mg/80 mg-Tablette enthält 535,8 mg Lactose-Monohydrat.
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
3. DARREICHUNGSFORM
Tablette.
Weiße bis gebrochen weiße, kapselförmige Tabletten mit der Aufschrift „311“, „312“, „313“ oder „315“ auf einer Seite.
4. KLINISCHE ANGABEN
4.1 Anwendungsgebiete
Prävention kardiovaskulärer Ereignisse
GOLTOR ist angezeigt zur Risikoreduktion von kardiovaskulären Ereignissen (siehe Abschnitt 5.1) bei Patienten mit koronarer Herzkrankheit (KHK) und akutem Koronarsyndrom in der Vorgeschichte, unabhängig von einer Vorbehandlung mit einem Statin.
Hypercholesterinämie
GOLTOR1 ist begleitend zu Diät angezeigt zur Anwendung bei Patienten mit primärer (heterozygoter familiärer und nicht familiärer) Hypercholesterinämie oder gemischter Hyperlipidämie, für die eine Therapie mit einem Kombinationspräparat geeignet ist:
• Patienten, bei denen eine Therapie mit einem Statin allein nicht ausreicht
• Patienten, die bereits mit einem Statin und Ezetimib behandelt werden Homozygote familiäre Hypercholesterinämie (HoFH)
GOLTOR ist begleitend zu Diät angezeigt zur Anwendung bei Patienten mit homozygoter familiärer Hypercholesterinämie. Die Patienten können weitere begleitende Therapien (wie LDL [low-density lipoprotein]-Apherese) erhalten.
4.2 Dosierung und Art der Anwendung
Dosierung
Hypercholesterinämie
Der Patient sollte eine geeignete lipidsenkende Diät einhalten, die er auch während der Therapie mit GOLTOR fortsetzen sollte.
Die Anwendung erfolgt oral. Der Dosierungsbereich von GOLTOR reicht von 10 mg/10 mg pro Tag bis zu 10 mg/80 mg pro Tag am Abend2. Die übliche Dosis beträgt 10 mg/20 mg oder 10 mg/40 mg pro Tag als Einzeldosis am Abend. Die 10 mg/80 mg-Dosis wird nur für Patienten mit schwerer Hypercholesterinämie und mit hohem Risiko für kardiovaskuläre Komplikationen empfohlen, die ihr Behandlungsziel mit einer niedrigeren Dosis nicht erreicht haben, und wenn zu erwarten ist, dass der Nutzen der Behandlung ihre potenziellen Risiken überwiegt (siehe Abschnitte 4.4 und 5.1). Bei Therapiebeginn oder bei einer Dosisänderung sind die LDL-Cholesterinwerte des Patienten, sein Risiko für die Entwicklung einer koronaren Herzkrankheit sowie sein Ansprechen auf eine bisherige lipidsenkende Therapie zu berücksichtigen.
Die Dosis von GOLTOR sollte individuell auf Basis der bekannten Wirksamkeit der verschiedenen Stärken von GOLTOR (siehe Abschnitt 5.1, Tabelle 4) sowie dem Ansprechen auf die bisherige lipidsenkende Therapie ausgewählt werden. Dosisanpassungen - falls erforderlich - sollten in Abständen von mindestens 4 Wochen durchgeführt werden. GOLTOR kann unabhängig von der Nahrungsaufnahme eingenommen werden. Die Tablette sollte nicht geteilt werden.
Patienten mit koronarer Herzkrankheit (KHK) und akutem Koronarsyndrom in der Vorgeschichte In der Studie zur Untersuchung der Risikoreduktion von kardiovaskulären Ereignissen (IMPROVEIT) betrug die Anfangsdosierung 10 mg/40 mg pro Tag am Abend. Die 10 mg/80 mg Dosierung wird nur empfohlen, wenn zu erwarten ist, dass der Nutzen der Behandlung die potentiellen Risiken überwiegt.
Homozygote familiäre Hypercholesterinämie
Die empfohlene Anfangsdosis für Patienten mit homozygoter familiärer Hypercholesterinämie beträgt GOLTOR 10 mg/40 mg pro Tag am Abend eingenommen. Die 10 mg/80 mg Dosierung wird nur empfohlen, wenn zu erwarten ist, dass der Nutzen der Behandlung die potenziellen Risiken überwiegt (siehe oben und die Abschnitte 4.3 und 4.4). GOLTOR kann bei diesen Patienten sowohl begleitend zu anderen lipidsenkenden Maßnahmen (z. B. LDL-Apherese) angewendet werden oder auch unabhängig davon, wenn solche Maßnahmen nicht zur Verfügung stehen.
Bei Patienten, die gleichzeitig Lomitapid und GOLTOR einnehmen, darf eine Dosis von GOLTOR 10 mg/40 mg pro Tag nicht überschritten werden (siehe Abschnitte 4.3, 4.4 und 4.5).
Gemeinsame Gabe mit anderen Arzneimitteln
Die Einnahme von GOLTOR sollte mindestens 2 Stunden vor oder mindestens 4 Stunden nach der Einnahme eines Anionenaustauschers erfolgen.
Bei Patienten, die gleichzeitig Amiodaron, Amlodipin, Verapamil oder Diltiazem mit GOLTOR einnehmen, sollte eine Dosis von GOLTOR 10 mg/20 mg pro Tag nicht überschritten werden (siehe Abschnitte 4.4 und 4.5).
Bei Patienten, die gleichzeitig Niacin in lipidsenkenden Dosen (> 1 g/Tag) mit GOLTOR einnehmen, sollte eine Dosis von GOLTOR 10 mg/20 mg pro Tag nicht überschritten werden (siehe Abschnitte 4.4 und 4.5).
Ältere Patienten
Für ältere Patienten ist keine Dosisanpassung erforderlich (siehe Abschnitt 5.2).
Kinder und Jugendliche
Die Behandlung ist von einem Spezialisten einzuleiten.
Jugendliche ab 10 Jahren (pubertärer Status: Jungen: Tanner-Stadium II und darüber; Mädchen: mindestens 1 Jahr nach der Menarche): Die klinische Erfahrung bei pädiatrischen und jugendlichen Patienten (im Alter von 10 bis 17 Jahren) ist begrenzt. Die empfohlene Anfangsdosis beträgt 10 mg/10 mg pro Tag am Abend. Der empfohlene Dosierungsbereich reicht von 10 mg/10 mg bis zu maximal 10 mg/40 mg pro Tag (siehe Abschnitte 4.4 und 5.2).
Kinder unter 10 Jahren: Aufgrund der unzureichenden Datenlage zur Sicherheit und Wirksamkeit wird die Behandlung mit GOLTOR für Kinder unter 10 Jahren nicht empfohlen (siehe Abschnitt 5.2). Die Erfahrung bei Kindern vor der Pubertät ist begrenzt.
Patienten mit eingeschränkter Leberfunktion
Für Patienten mit leichter Einschränkung der Leberfunktion (Child-Pugh-Score 5-6) ist keine Dosisanpassung erforderlich. Für Patienten mit mäßiger (Child-Pugh-Score 7-9) oder schwerer (Child-Pugh-Score > 9) Einschränkung der Leberfunktion wird die Behandlung mit GOLTOR nicht empfohlen (siehe Abschnitte 4.4 und 5.2).
Patienten mit eingeschränkter Nierenfunktion
Bei Patienten mit leichter Einschränkung der Nierenfunktion (glomeruläre Filtrationsrate > 60 ml/min/1,73 m2) ist in der Regel keine Dosisanpassung erforderlich. Bei Patienten mit chronischer Nierenerkrankung mit einer glomerulären Filtrationsrate von < 60 ml/min/1,73 m2 beträgt die empfohlene GOLTOR Dosis 10 mg/20 mg, einmal täglich am Abend (siehe Abschnitte 4.4, 5.1 und 5.2). Höhere Dosen sollten mit Vorsicht angewendet werden.
Art der Anwendung
GOLTOR wird oral eingenommen. GOLTOR kann als Einzeldosis am Abend eingenommen werden.
4.3 Gegenanzeigen
Überempfindlichkeit gegen die Wirkstoffe, oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Schwangerschaft und Stillzeit (siehe Abschnitt 4.6).
Aktive Lebererkrankung oder unklare und andauernde Erhöhung der Serum-Transaminasen.
Gleichzeitige Anwendung von potenten CYP3A4-Inhibitoren (Substanzen, welche die AUC mindestens um ca. das 5-Fache erhöhen) (z. B. Itraconazol, Ketoconazol, Posaconazol, Voriconazol, Erythromycin, Clarithromycin, Telithromycin, HIV-Protease-Inhibitoren [z. B. Nelfinavir], Boceprevir, Telaprevir, Nefazodon und Arzneimittel, die Cobicistat enthalten) (siehe Abschnitte 4.4 und 4.5).
Gleichzeitige Anwendung von Gemfibrozil, Ciclosporin oder Danazol (siehe Abschnitte 4.4 und 4.5).
Gleichzeitige Anwendung von Lomitapid und GOLTOR in Dosen von mehr als 10 mg/40 mg bei Patienten mit homozygoter familiärer Hypercholesterinämie (HoFH) (siehe Abschnitte 4.2, 4.4 und 4.5).
4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Mvopathie/Rhabdomvolvse
Nach Markteinführung von Ezetimib wurden Fälle von Myopathie und Rhabdomyolyse berichtet. Die meisten Patienten, die eine Rhabdomyolyse entwickelten, nahmen gleichzeitig mit Ezetimib ein Statin ein. Jedoch wurde eine Rhabdomyolyse sehr selten unter Monotherapie mit Ezetimib sowie sehr selten
nach Zugabe von Ezetimib zu anderen Arzneimitteln berichtet, die bekanntermaßen mit einem erhöhten Rhabdomyolyserisiko in Verbindung stehen.
GOLTOR enthält Simvastatin. Wie andere HMG-CoA-Reduktase-Inhibitoren ruft Simvastatin gelegentlich eine Myopathie hervor, die sich in Muskelschmerzen, -empfindlichkeit oder -schwäche verbunden mit Erhöhungen der Kreatinkinase (CK) (> das Zehnfache des oberen Normwertes) äußert. Manchmal manifestiert sich die Myopathie als Rhabdomyolyse mit oder ohne akutem Nierenversagen aufgrund von Myoglobinurie, sehr selten mit tödlichem Ausgang. Das Risiko einer Myopathie ist bei hoher HMG-CoA-Reduktase-Inhibitoraktivität im Plasma erhöht.
Das Risiko für eine Myopathie/Rhabdomyolyse ist für Simvastatin wie für andere HMG-CoA-Reduktase-Inhibitoren dosisabhängig. In einer Datenbank für klinische Studien wurden 41.413 Patienten erfasst, die mit Simvastatin behandelt wurden, darunter 24.747 (ca. 60 %) in Studien mit einer medianen Beobachtungsdauer von mindestens 4 Jahren. Die Myopathiehäufigkeit lag annähernd bei 0,03 % unter 20 mg, bei 0,08 % unter 40 mg und bei 0,61 % unter 80 mg Simvastatin pro Tag. In diesen Studien wurden die Patienten sorgfältig überwacht und einige interagierende Arzneimittel wurden ausgeschlossen.
In einer klinischen Studie erhielten Patienten mit einem Myokardinfarkt in der Vorgeschichte 80 mg Simvastatin pro Tag (mittlere Beobachtungsdauer 6,7 Jahre). Die Myopathiehäufigkeit lag bei ca.
1.0 % im Vergleich zu 0,02 % bei Patienten unter 20 mg pro Tag. Etwa die Hälfte dieser Myopathiefalle ereignete sich im ersten Jahr der Behandlung. Die Myopathiehäufigkeit in den folgenden Jahren lag jeweils bei ca. 0,1 % (siehe Abschnitte 4.8 und 5.1).
Das Myopathierisiko ist für Patienten unter GOLTOR 10 mg/80 mg im Vergleich zu anderen Statinbasierten Therapien mit ähnlicher LDL-senkender Wirksamkeit größer. Demzufolge sollte GOLTOR 10 mg/80 mg nur Patienten mit schwerer Hypercholesterinämie und mit hohem Risiko für kardiovaskuläre Komplikationen gegeben werden, die ihr Behandlungsziel mit niedrigeren Dosierungen nicht erreicht haben, und wenn der zu erwartende Nutzen die potenziellen Risiken übersteigt. Patienten unter GOLTOR 10 mg/80 mg, die zusätzlich ein anderes, damit wechselwirkendes, Arzneimittel benötigen, sollten auf eine niedrigere GOLTOR Dosis oder auf eine alternative Statintherapie eingestellt werden, welche ein geringeres Potenzial für Arzneimittelwechselwirkungen hat (siehe nachfolgend unter Maßnahmen zur Verringerung des Myopathierisikos aufgrund von Arzneimittelwechselwirkungen sowie die Abschnitte 4.2, 4.3 und 4.5).
In der IMPROVE-IT-Studie (IMProved Reduction of Outcomes: Vytorin Efficacy International Trial) erhielten 18.144 Patienten mit koronarer Herzkrankheit und akutem Koronarsyndrom in der Vorgeschichte randomisiert entweder einmal täglich Ezetimib/Simvastatin 10 mg/40 mg (n = 9.067) oder einmal täglich Simvastatin 40 mg (n = 9.077). Während der medianen Nachbeobachtung von
6.0 Jahren betrug die Inzidenz einer Myopathie 0,2 % in der GOLTOR-Gruppe und 0,1 % in der Simvastatin-Monotherapie-Gruppe. Myopathie war definiert als Muskelschwäche oder Muskelschmerzen ungeklärter Ursache mit einer Erhöhung des Serumkreatinins (CK) um das > 10Fache des oberen Normwertes [ULN] oder zwei aufeinanderfolgenden Erhöhungen des Serumkreatinins (CK) um das > 5 - < 10-Fache des oberen Normwertes [ULN]. Die Inzidenz einer Rhabdomyolyse betrug 0,1 % in der GOLTOR-Gruppe und 0,2 % in der Simvastatin-Monotherapie-Gruppe. Rhabdomyolyse war definiert als Muskelschwäche oder Muskelschmerzen ungeklärter Ursache mit einer Erhöhung des Serumkreatinins (CK) um das > 10-Fache des oberen Normwertes [ULN] mit dem Nachweis einer Nierenschädigung oder zwei aufeinanderfolgenden Erhöhungen des Serumkreatinins (CK) um das > 5 - < 10-Fache des oberen Normwertes [ULN] mit Nachweis einer Nierenschädigung oder mit einem Serumkreatinin (CK) von > 10.000 IU/l ohne Nachweis einer Nierenschädigung (siehe Abschnitt 4.8).
In einer klinischen Studie, in der mehr als 9.000 Patienten mit chronischer Nierenerkrankung randomisiert entweder GOLTOR 10 mg/20 mg einmal täglich (n = 4.650) oder Plazebo (n = 4.620) (mediane Verlaufsbeobachtung 4,9 Jahre) erhielten, betrug die Inzidenz für Myopathien 0,2 % unter GOLTOR bzw. 0,1 % unter Plazebo (siehe Abschnitt 4.8).
Im Rahmen einer klinischen Studie (mediane Nachbeobachtung 3,9 Jahre), bei der Patienten mit hohem kardiovaskulären Risiko mit Simvastatin 40 mg/Tag behandelt wurden, lag die Inzidenz für Myopathien bei nicht-chinesischen Patienten (n = 7.367) bei ca. 0,05 % im Vergleich zu 0,24 % bei chinesischen Patienten (n = 5.468). Obwohl im Rahmen dieser klinischen Studie ausschließlich chinesische Patienten als einzige asiatische Population untersucht und ausgewertet wurden, sollte GOLTOR generell nur mit Vorsicht bei asiatischen Patienten und in der niedrigsten erforderlichen Dosis verordnet werden.
Reduzierte Funktion von Transporterproteinen
Die verminderte Funktion des hepatischen OATP-Transportproteins kann die systemische Exposition von Simvastatinsäure sowie das Risiko für Myopathie und Rhabdomyolyse erhöhen. Die verminderte Funktion kann als Ergebnis einer Hemmung durch interagierende Arzneimittel (z. B. Ciclosporin) entstehen oder bei Patienten auftreten, die Träger des SLCO1B1-c.521T>C-Genotyps sind.
Patienten, die das Allel c.521T>C des SLCO1B1-Gens tragen, das ein weniger aktives OATP1B1-Protein kodiert, haben eine erhöhte systemische Exposition von Simvastatinsäure sowie ein erhöhtes Myopathierisiko. Das Risiko einer durch hochdosiertes Simvastatin (80 mg) bedingten Myopathie liegt ohne Gentest im Allgemeinen bei 1 %. Basierend auf den Ergebnissen der SEARCH-Studie haben mit 80 mg behandelte Träger des homozygoten C-Allels (auch CC genannt) ein 15 %iges Risiko für eine Myopathie innerhalb eines Jahres, während das Risiko bei Trägern des heterozygoten C-Allels (CT) bei 1,5 % liegt. Patienten mit dem häufigsten Genotyp (TT) haben diesbezüglich ein Risiko von 0,3 % (siehe Abschnitt 5.2). Sofern verfügbar, sollten eine Genotypisierung bezüglich des Vorliegens des C-Allels als Teil der Nutzen-Risiko-Bewertung bei einzelnen Patienten vor einer Verordnung von Simvastatin 80 mg in Betracht gezogen sowie hohe Dosen bei identifizierten Trägern des CC-Genotyps vermieden werden. Die Abwesenheit dieses Gens bei der Genotypisierung schließt allerdings nicht aus, dass eine Myopathie auftreten kann.
Messungen der Kreatinkinase (CK)
Die Kreatinkinase (CK) sollte nicht nach körperlicher Anstrengung oder bei Vorliegen anderer plausibler Ursachen für eine CK-Erhöhung gemessen werden, da dies eine Interpretation der Werte erschwert. Wenn die Ausgangswerte der CK signifikant erhöht sind (> das Fünffache des oberen Normwertes), sollte die Messung nach 5-7 Tagen wiederholt werden, um die Ergebnisse zu bestätigen.
Vor Beginn der Therapie
Alle Patienten, die auf GOLTOR eingestellt werden oder deren Dosis von GOLTOR erhöht wird, sollten über das Risiko einer Myopathie aufgeklärt und aufgefordert werden, unklare Muskelschmerzen, -empfindlichkeit oder -schwäche umgehend mitzuteilen.
Bei Patienten mit Risikofaktoren für eine Rhabdomyolyse ist Vorsicht angebracht. Um einen Ausgangswert als Referenz festzustellen, sollten in folgenden Situationen vor Behandlungsbeginn Bestimmungen der CK durchgeführt werden:
• ältere Patienten (> 65 Jahre alt)
• weibliche Patienten
• Nierenfunktionsstörung
• unbehandelte Hypothyreose
• hereditäre Muskelerkrankungen in der eigenen oder in der Familienanamnese
• muskuläre Symptomatik unter Behandlung mit Statinen oder Fibraten in der Anamnese
• Alkoholmissbrauch
In solchen Fällen wird eine sorgfältige Nutzen-Risiko-Abwägung der Behandlung empfohlen. Die betroffenen Patienten sollten engmaschig überwacht werden. Bei Patienten, bei denen bereits eine Muskelerkrankung unter Behandlung mit Fibraten oder Statinen aufgetreten ist, sollte die Behandlung mit allen Produkten, die ein Statin enthalten (wie auch GOLTOR), nur mit Vorsicht begonnen werden. Wenn die CK-Ausgangswerte signifikant erhöht sind (> das Fünffache des oberen Normwertes), sollte nicht mit der Therapie begonnen werden.
Im Behandlungsverlauf
Wenn während der Behandlung mit GOLTOR Muskelschmerzen, -schwäche oder Krämpfe auftreten, sollten die CK-Werte bestimmt werden. Wenn die CK-Werte ohne körperliche Anstrengung signifikant erhöht sind (> das Fünffache des oberen Normwertes), ist die Therapie zu beenden. Sollte die muskuläre Symptomatik schwerwiegend sein und tägliche Beeinträchtigungen verursachen, kann erwogen werden, die Behandlung abzusetzen, auch wenn die CK-Werte weniger als auf das Fünffache des oberen Normwertes erhöht sind. Bei Verdachtsdiagnose einer Myopathie anderer Ursache sollte die Therapie beendet werden.
In sehr seltenen Fällen wurde während oder nach der Behandlung mit einigen Statinen über eine immunvermittelte nekrotisierende Myopathie (immune-mediated necrotizing myopathy; IMNM) berichtet. Die klinischen Charakteristika einer IMNM sind persistierende proximale Muskelschwäche und erhöhte Serum-Kreatinkinase-Werte, die trotz Absetzen der Behandlung mit Statinen fortbestehen (siehe Abschnitt 4.8).
Wenn die Symptome verschwinden und die CK-Werte zurückgehen, kann die erneute Behandlung mit GOLTOR oder mit einem alternativen statinhaltigen Produkt in der jeweils niedrigsten Dosis und bei engmaschiger Überwachung in Erwägung gezogen werden.
Eine erhöhte Myopathierate wurde bei Patienten beobachtet, die auf die 80-mg-Dosis Simvastatin eingestellt wurden (siehe Abschnitt 5.1). Es wird empfohlen, die CK-Werte regelmäßig zu überwachen, was bei der Identifizierung von Myopathien ohne klinische Symptome von Nutzen sein könnte. Eine derartige Überwachung kann jedoch eine Myopathie nicht mit Gewissheit verhindern.
Die Therapie mit GOLTOR sollte einige Tage vor größeren geplanten chirurgischen Eingriffen sowie bei Eintritt eines akuten ernsten Krankheitsbildes bzw. Notwendigkeit von chirurgischen Maßnahmen vorübergehend unterbrochen werden.
Maßnahmen zur Verringerung des Myopathierisikos aufgrund von Arzneimittelwechselwirkungen (siehe auch Abschnitt 4.5)
Das Risiko einer Myopathie und Rhabdomyolyse ist signifikant erhöht bei gleichzeitiger Anwendung von GOLTOR mit potenten Inhibitoren von CYP3A4 (wie z. B. Itraconazol, Ketoconazol, Posaconazol, Voriconazol, Erythromycin, Clarithromycin, Telithromycin, HIV-Protease-Inhibitoren [z. B. Nelfinavir], Boceprevir, Telaprevir, Nefazodon und Arzneimittel, die Cobicistat enthalten) sowie mit Ciclosporin, Danazol und Gemfibrozil. Die gleichzeitige Anwendung von GOLTOR mit Arzneimitteln mit diesen Wirkstoffen ist kontraindiziert (siehe Abschnitt 4.3).
Aufgrund des in GOLTOR enthaltenen Wirkstoffs Simvastatin ist das Risiko einer Myopathie und Rhabdomyolyse ebenfalls erhöht bei gleichzeitiger Anwendung mit anderen Fibraten, Niacin in lipidsenkenden Dosen (> 1 g/Tag) oder bei gleichzeitiger Therapie von Amiodaron, Amlodipin, Verapamil oder Diltiazem mit bestimmten Dosen von GOLTOR (siehe Abschnitte 4.2 und 4.5). Das Risiko einer Myopathie einschließlich einer Rhabdomyolyse kann durch die gleichzeitige Anwendung von Fusidinsäure und GOLTOR erhöht werden. Bei Patienten mit homozygoter familiärer Hypercholesterinämie (HoFH) kann dieses Risiko durch die gemeinsame Anwendung von GOLTOR mit Lomitapid erhöht sein (siehe Abschnitt 4.5).
Folglich ist hinsichtlich der CYP3A4-Inhibitoren eine gleichzeitige Anwendung von GOLTOR mit Itraconazol, Ketoconazol, Posaconazol, Voriconazol, HIV-Protease-Inhibitoren (z. B. Nelfinavir), Boceprevir, Telaprevir, Erythromycin, Clarithromycin, Telithromycin, Nefazodon und Arzneimitteln, die Cobicistat enthalten, kontraindiziert (siehe Abschnitte 4.3 und 4.5). Falls eine Behandlung mit potenten CYP3A4-Inhibitoren (Substanzen, welche die AUC mindestens um ca. das 5-Fache erhöhen) unabdingbar ist, muss die Therapie mit GOLTOR während der Behandlungsdauer unterbrochen werden (und die Anwendung eines alternativen Statins in Erwägung gezogen werden). Außerdem ist Vorsicht angebracht, wenn GOLTOR mit bestimmten anderen weniger potenten CYP3A4-Inhibitoren kombiniert wird: Fluconazol, Verapamil und Diltiazem (siehe Abschnitte 4.2 und 4.5). Genuss von Grapefruitsaft sollte während der Behandlung mit GOLTOR vermieden werden.
Simvastatin darf nicht zusammen mit Fusidinsäure gegeben werden. Es wurde über das Auftreten von Rhabdomyolyse (einschließlich einiger mit Todesfolge) bei Patienten berichtet, welche diese Kombination erhielten (siehe Abschnitt 4.5). Sofern die systemische Gabe von Fusidinsäure bei Patienten als essenziell erachtet wird, ist die Statintherapie während der gesamten Behandlungsdauer mit Fusidinsäure abzusetzen. Die Patienten sollten darüber informiert werden, sich umgehend an einen Arzt zu wenden, wenn sie Anzeichen von Muskelschwäche, -schmerzen oder -empfindlichkeit bemerken.
Die Statintherapie kann 7 Tage nach der letzten Dosis Fusidinsäure fortgesetzt werden. Sofern in Ausnahmefällen eine längere systemische Gabe von Fusidinsäure notwendig ist, wie z. B. zur Behandlung von schweren Infektionen, sollte eine gemeinsame Gabe von GOLTOR mit Fusidinsäure nur im Einzelfall unter engmaschiger medizinischer Überwachung in Betracht gezogen werden.
Die Kombination von höheren GOLTOR Dosen als 10 mg/20 mg pro Tag mit lipidsenkenden Dosen von Niacin (> 1 g/Tag) sollte vermieden werden, sofern der klinische Nutzen das erhöhte Risiko einer Myopathie nicht überwiegt (siehe Abschnitte 4.2 und 4.5).
Die Kombination von HMG-CoA-Reduktase-Hemmern und Niacin (Nicotinsäure) in lipidsenkenden Dosen (> 1 g/Tag) wurde mit selten auftretenden Fällen von Myopathie/Rhabdomyolyse in Verbindung gebracht; die alleinige Gabe jeder dieser Einzelsubstanzen kann bereits eine Myopathie auslösen.
Im Rahmen einer klinischen Studie (mediane Nachbeobachtung 3,9 Jahre) bei Patienten mit hohem kardiovaskulären Risiko und gut eingestellten LDL-Cholesterinspiegeln, die Simvastatin 40 mg/Tag mit oder ohne Ezetimib 10 mg erhielten, wurde durch Zugabe von Niacin (Nicotinsäure) in lipidsenkenden Dosen (> 1 g/Tag) kein zusätzlicher Nutzen im Hinblick auf das kardiovaskuläre Outcome beobachtet. Ärzte, die eine Kombinationstherapie mit Simvastatin und Niacin (Nicotinsäure) in lipidsenkenden Dosen (> 1 g/Tag) oder niacinhaltigen Präparaten in Erwägung ziehen, sollten demzufolge eine sorgfältige Nutzen-Risiko-Analyse durchführen und die Patienten sorgfältig auf jegliche Anzeichen und Symptome von Schmerzen, Empfindlichkeit oder Schwäche der Muskulatur überwachen, insbesondere in den ersten Monaten der Behandlung sowie bei Dosiserhöhung einer oder beider Einzelsubstanzen.
Außerdem lag bei dieser Studie die Inzidenz für Myopathien bei chinesischen Patienten unter Simvastatin 40 mg oder Ezetimib/Simvastatin 10 mg/40 mg bei ca. 0,24 % im Vergleich zu 1,24 % bei chinesischen Patienten unter Simvastatin 40 mg oder Ezetimib/Simvastatin 10 mg/40 mg, die zusätzlich mit Nicotinsäure/Laropiprant 2.000 mg/40 mg mit veränderter Wirkstofffreisetzung behandelt wurden. Obwohl im Rahmen dieser klinischen Studie ausschließlich chinesische Patienten als einzige asiatische Population untersucht und ausgewertet wurden, und die Inzidenz für Myopathien bei chinesischen im Vergleich zu nicht-chinesischen Patienten höher ist, wird die gemeinsame Anwendung von GOLTOR mit lipidsenkenden Dosen (> 1 g/Tag) von Niacin (Nicotinsäure) generell bei asiatischen Patienten nicht empfohlen.
Der Wirkstoff Acipimox ist strukturell mit Niacin verwandt. Obwohl Acipimox nicht untersucht wurde, könnten die Risiken für myotoxische Effekte ähnlich wie bei Niacin sein.
Die Kombination von höheren Dosen als GOLTOR 10 mg/20 mg pro Tag mit Amiodaron, Amlodipin, Verapamil oder Diltiazem sollte vermieden werden. Bei Patienten mit homozygoter familiärer Hypercholesterinämie (HoFH), ist die gemeinsame Anwendung von GOLTOR in Dosen von mehr als 10 mg/40 mg mit Lomitapid zu vermeiden (siehe Abschnitte 4.2, 4.3 und 4.5).
Patienten, die gleichzeitig mit GOLTOR (vor allem hohe GOLTOR Dosierungen) andere Arzneimittel einnehmen, die bei therapeutischer Dosierung moderate CYP3A4-Inhibitoren sind, könnten ein erhöhtes Myopathierisiko haben. Sofern GOLTOR gleichzeitig mit einem moderaten CYP3A4-Inhibitor (Substanzen, welche die AUC um ca. das 2- bis 5-Fache erhöhen) gegeben wird, kann eine Dosisanpassung erforderlich sein. Bei bestimmten moderaten CYP3A4-Inhibitoren wie z. B.
Diltiazem wird empfohlen eine Tageshöchstdosis von GOLTOR 10 mg/20 mg nicht zu überschreiten (siehe Abschnitt 4.2).
Sicherheit und Wirksamkeit von GOLTOR zusammen mit Fibraten wurden nicht untersucht. Das Risiko für eine Myopathie ist erhöht, wenn Simvastatin gemeinsam mit Fibraten (insbesondere mit Gemfibrozil) angewendet wird. Daher ist die gemeinsame Anwendung von GOLTOR und Gemfibrozil kontraindiziert (siehe Abschnitt 4.3). Ebenfalls wird daher die gleichzeitige Anwendung mit anderen Fibraten nicht empfohlen (siehe Abschnitt 4.5).
Leberenzyme
In kontrollierten klinischen Studien zur Koadministration wurden bei Patienten, die Simvastatin zusammen mit Ezetimib erhielten, Erhöhungen der Transaminasenwerte (> dem Dreifachen des oberen Normwertes [ULN] in Folge) beobachtet (siehe Abschnitt 4.8).
In der IMPROVE-IT-Studie (IMProved Reduction of Outcomes: Vytorin Efficacy International Trial) erhielten 18.144 Patienten mit koronarer Herzkrankheit und akutem Koronarsyndrom in der Vorgeschichte randomisiert entweder einmal täglich Ezetimib/Simvastatin 10 mg/40 mg (n = 9.067) oder einmal täglich Simvastatin 40 mg (n = 9.077). Während der medianen Nachbeobachtung von
6,0 Jahren betrug die Inzidenz einer konsekutiven Erhöhung der Transaminasenwerte (> dem Dreifachen des oberen Normwertes [ULN]) 2,5 % in der GOLTOR-Gruppe und 2,3 % in der Simvastatin-Monotherapie-Gruppe (siehe Abschnitt 4.8).
In einer kontrollierten klinischen Studie, in der mehr als 9.000 Patienten mit chronischer Nierenerkrankung randomisiert entweder GOLTOR 10 mg/20 mg täglich (n = 4.650) oder Plazebo (n = 4.620) (mediane Verlaufsbeobachtung 4,9 Jahre) erhielten, betrug die Inzidenz einer konsekutiven Erhöhung der Transaminasenwerte (> dem Dreifachen des oberen Normwertes [ULN]) 0,7 % unter GOLTOR und 0,6 % unter Plazebo (siehe Abschnitt 4.8).
Leberfunktionstests werden vor Beginn der Behandlung mit GOLTOR und danach immer, wenn klinisch angezeigt, empfohlen. Bei Patienten, die auf eine Dosis von 10 mg/80 mg eingestellt wurden, sollte eine zusätzliche Bestimmung vor der Dosiserhöhung, drei Monate nach Dosiserhöhung auf 10 mg/80 mg und danach in regelmäßigen Abständen (z. B. halbjährlich) im ersten Behandlungsjahr erfolgen. Besondere Aufmerksamkeit sollte denjenigen Patienten gelten, die während der Therapie erhöhte Transaminasenspiegel entwickeln; bei diesen Patienten sollten die Bestimmungen umgehend wiederholt und häufiger durchgeführt werden. Sollten die Transaminasenerhöhungen weiter fortschreiten, insbesondere wenn sie bis zum Dreifachen der oberen Normgrenze ansteigen und persistieren, sollte das Arzneimittel abgesetzt werden. Es sollte beachtet werden, dass ALT aus dem Muskelgewebe freigesetzt werden kann. Daher kann ein Anstieg von ALT mit CK ein Hinweis auf eine Myopathie sein (siehe vorher unter Myopathie/Rhabdomyolyse).
Nach Markteinführung wurde bei Patienten, die Statine einschließlich Simvastatin einnahmen, selten über Leberversagen mit teils tödlichem Ausgang berichtet. Sofern während der Behandlung mit GOLTOR schwerwiegende Beeinträchtigungen der Leber mit entsprechender klinischer Symptomatik und/oder Hyperbilirubinämie oder Ikterus auftreten, muss die Behandlung unverzüglich abgebrochen werden. Wenn keine andere Ursache feststellbar ist, darf die Behandlung mit GOLTOR nicht fortgesetzt werden.
GOLTOR sollte mit Vorsicht bei denjenigen Patienten eingesetzt werden, die in erheblichem Maße Alkohol zu sich nehmen.
Einschränkung der Leberfunktion
Aufgrund fehlender Daten zu Auswirkungen einer erhöhten Exposition von Ezetimib bei Patienten mit mäßiger oder schwerer Einschränkung der Leberfunktion wird GOLTOR für diese Patienten nicht empfohlen (siehe Abschnitt 5.2).
Diabetes mellitus
Es gibt Hinweise darauf, dass Statine als Substanzklasse den Blutzuckerspiegel erhöhen und bei manchen Patienten, die ein hohes Risiko für die Entwicklung eines zukünftigen Diabetes mellitus haben, eine Hyperglykämie hervorrufen können, die eine adäquate Diabetesbehandlung erfordert. Dieses Risiko wird jedoch von der Reduktion des vaskulären Risikos durch Statine aufgewogen und sollte daher nicht zu einem Abbruch der Statinbehandlung führen. In Übereinstimmung mit nationalen Richtlinien sollten Risikopatienten (Nüchternblutzucker von 5,6 bis 6,9 mmol/l, BMI > 30 kg/m2, erhöhte Triglyzeridwerte, Hypertonie) sowohl klinisch als auch in Bezug auf die relevanten Laborwerte überwacht werden.
Kinder und Jugendliche
Wirksamkeit und Sicherheit von Ezetimib zusammen mit Simvastatin wurden bei Patienten im Alter von 10 bis 17 Jahren mit heterozygoter familiärer Hypercholesterinämie in einer kontrollierten klinischen Studie mit heranwachsenden Jungen (Tanner-Stadium II oder darüber) und Mädchen (mindestens 1 Jahr nach der Menarche) untersucht.
In dieser begrenzten kontrollierten Studie war im Allgemeinen keine Auswirkung auf Wachstum oder sexuelle Entwicklung bei den heranwachsenden Jungen oder Mädchen erkennbar, auch keine Auswirkung auf die Länge des Menstruationszyklus der Mädchen. Jedoch wurde die Auswirkung von Ezetimib über einen längeren Zeitraum als 33 Wochen auf Wachstum und sexuelle Entwicklung nicht untersucht (siehe Abschnitte 4.2 und 4.8).
Sicherheit und Wirksamkeit von Ezetimib in Kombination mit Simvastatin in Dosen über 40 mg pro Tag wurden bei pädiatrischen Patienten im Alter von 10 bis 17 Jahren nicht untersucht.
Ezetimib wurde nicht bei Patienten unter 10 Jahren oder bei Mädchen vor der Menarche untersucht (siehe Abschnitte 4.2 und 4.8).
Die Langzeitwirkung einer Therapie mit Ezetimib bei Patienten unter 17 Jahren auf die Reduktion von Morbidität und Mortalität im Erwachsenenalter wurde nicht untersucht.
Fibrate
Sicherheit und Wirksamkeit von Ezetimib zusammen mit Fibraten wurden nicht untersucht (siehe oben und die Abschnitte 4.3 und 4.5).
Antikoagulanzien
Bei Zugabe von GOLTOR zu Warfarin, einem anderen Cumarin-Antikoagulans oder Fluindion ist die „International Normalized Ratio“ (INR) entsprechend zu überwachen (siehe Abschnitt 4.5).
Interstitielle Lungenkrankheit
Bei einigen Statinen, einschließlich Simvastatin, wurden besonders bei Langzeittherapie Fälle einer interstitiellen Lungenkrankheit berichtet (siehe Abschnitt 4.8). Die auftretenden Beschwerden können dabei Dyspnoe, unproduktiven Husten und allgemeine Gesundheitsstörungen (Erschöpfung, Gewichtsverlust und Fieber) einschließen. Wenn vermutet wird, dass ein Patient eine interstitielle Lungenkrankheit entwickelt hat, sollte die Statintherapie abgebrochen werden.
Sonstiger Bestandteil
Patienten mit der seltenen hereditären Galactose-Intoleranz, Lactase-Mangel oder Glucose-Galactose-Malabsorption sollten diese Arzneimittel nicht einnehmen.
4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Pharmakodynamische Wechselwirkungen
Wechselwirkungen mit lipidsenkenden Arzneimitteln, die bei Monotherapie eine Myopathie verursachen können
Das Risiko einer Myopathie einschließlich einer Rhabdomyolyse ist während gemeinsamer Gabe von Simvastatin mit Fibraten erhöht. Mit Gemfibrozil besteht außerdem eine pharmakokinetische Interaktion für Simvastatin, die zu erhöhten Plasmakonzentrationen von Simvastatin führt (siehe unten Pharmakokinetische Wechselwirkungen und die Abschnitte 4.3 und 4.4). Die Kombination von Simvastatin und Niacin (Nicotinsäure) in lipidsenkenden Dosen (> 1 g/Tag) wurde mit selten auftretenden Fällen von Myopathie/Rhabdomyolyse in Verbindung gebracht (siehe Abschnitt 4.4).
Fibrate können die Cholesterinausscheidung über die Galle erhöhen und so zu Cholelithiasis führen. In einer präklinischen Studie an Hunden erhöhte Ezetimib Cholesterin in der Galle (siehe Abschnitt 5.3). Auch wenn die Bedeutung dieser präklinischen Befunde für den Menschen unbekannt ist, wird die gemeinsame Gabe von GOLTOR mit Fibraten nicht empfohlen (siehe Abschnitt 4.4).
Pharmakokinetische Wechselwirkungen
Verordnungsempfehlungen zu interagierenden Arzneimitteln sind in der unten stehenden Tabelle (siehe Tabelle 1) zusammengefasst (weitere Details sind im Text erläutert; siehe auch Abschnitte 4.2, 4.3 und 4.4).
Tabelle 1:
Mit einem erhöhten Risiko für eine Myopathie/Rhabdomyolyse assoziierte Arzneimittelwechselwirkungen
|
Interagierende Stoffe |
Verordnungsempfehlungen |
|
Potente CYP3A4-Inhibitoren, wie z. B. | |
|
Itraconazol |
Gleichzeitige Anwendung mit GOLTOR ist |
|
Ketoconazol Posaconazol Voriconazol Erythromycin Clarithromycin Telithromycin HIV-Protease-Inhibitoren (z. B. Nelfinavir) Boceprevir Telaprevir Nefazodon Cobicistat Ciclosporin Danazol Gemfibrozil |
kontraindiziert |
|
Andere Fibrate Fusidinsäure |
Anwendung mit GOLTOR nicht empfohlen |
|
Niacin (Nicotinsäure) (> 1 g/Tag) |
Anwendung mit GOLTOR bei asiatischen Patienten nicht empfohlen |
|
Amiodaron |
Eine Dosis von 10 mg/20 mg GOLTOR pro |
|
Amlodipin Verapamil Diltiazem Niacin (> 1 g/Tag) |
Tag nicht überschreiten |
|
Lomitapid |
Bei Patienten mit homozygoter familiärer Hypercholesterinämie (HoFH) eine Dosis von 10 mg/40 mg GOLTOR pro Tag nicht überschreiten |
|
Grapefruitsaft |
Während der Behandlung mit GOLTOR Grapefruitsaft vermeiden |
Wirkungen anderer Arzneimittel auf GOLTOR GOLTOR
Niacin: In einer Studie mit 15 gesunden Erwachsenen verursachte die gleichzeitige Anwendung mit GOLTOR (7 Tage 10 mg/20 mg täglich) eine geringe Erhöhung der mittleren AUCs von Niacin (22 %) und Nikotinursäure (19 %), die als „NIASPAN Retardtabletten“ (2 Tage 1.000 mg und 5 Tage
2.000 mg nach einem fettarmen Frühstück) gegeben wurden. In derselben Studie erhöhte gleichzeitig gegebenes NIASPAN geringfügig die mittleren AUCs von Ezetimib (9 %), Gesamt-Ezetimib (26 %), Simvastatin (20 %) und Simvastatinsäure (35 %) (siehe Abschnitte 4.2 und 4.4).
Arzneimittelwechselwirkungsstudien mit höheren Dosen Simvastatin wurden nicht durchgeführt.
Ezetimib
Antazida: Die gleichzeitige Anwendung von Antazida verminderte die Resorptionsrate von Ezetimib, beeinflusste aber nicht die Bioverfügbarkeit von Ezetimib. Der verminderten Resorptionsrate wird keine klinische Bedeutung beigemessen.
Colestyramin: Die gleichzeitige Anwendung von Colestyramin verkleinerte die mittlere Fläche unter der Kurve (AUC) von Gesamt-Ezetimib (Ezetimib und glukuronidiertes Ezetimib) um ca. 55 %. Die gesteigerte Senkung des LDL-Cholesterins durch Hinzufügen von GOLTOR zu Colestyramin könnte durch diese Interaktion vermindert werden (siehe Abschnitt 4.2).
Ciclosporin: In einer Studie mit acht Patienten, die nach einer Nierentransplantation mit einer Kreatinin-Clearance > 50 ml/min stabil auf eine Ciclosporin-Dosis eingestellt waren, war nach Gabe einer Einzeldosis von 10 mg Ezetimib die mittlere AUC von Gesamt-Ezetimib 3,4fach vergrößert (Bereich von 2,3- bis 7,9fach) verglichen mit einer gesunden Kontrollpopulation einer anderen Studie (n = 17) unter Ezetimib allein. In einer weiteren Studie wies ein Patient nach einer Nierentransplantation mit schwerer Einschränkung der Nierenfunktion, der Ciclosporin und zahlreiche andere Arzneimittel erhielt, eine 12fach größere Gesamt-Ezetimib-Exposition auf im Vergleich zu den anderen Kontrollpersonen unter Ezetimib allein. In einer zweiphasigen Crossover-Studie mit 12 gesunden Probanden führte die tägliche Anwendung von 20 mg Ezetimib über 8 Tage mit einer Einzeldosis von 100 mg Ciclosporin an Tag 7 zu einer mittleren 15%igen Vergrößerung der AUC von Ciclosporin (Bereich von 10%iger Verkleinerung bis 51%iger Vergrößerung) verglichen mit einer Einzeldosis von 100 mg Ciclosporin allein. Es wurden keine kontrollierten Studien über die Veränderung der Ciclosporin-Exposition durch die gemeinsame Anwendung mit Ezetimib bei Patienten nach einer Nierentransplantation durchgeführt. Die gleichzeitige Anwendung von GOLTOR und Ciclosporin ist kontraindiziert (siehe Abschnitt 4.3).
Fibrate: Die gleichzeitige Anwendung von Fenofibrat oder Gemfibrozil erhöhte die Konzentration von Gesamt-Ezetimib auf das ca. 1,5- bzw. 1,7-Fache. Auch wenn diesen Erhöhungen keine klinische Bedeutung beigemessen wird, ist die gleichzeitige Gabe von GOLTOR mit Gemfibrozil kontraindiziert (siehe Abschnitt 4.3). Ebenfalls wird die gleichzeitige Gabe mit anderen Fibraten nicht empfohlen (siehe Abschnitt 4.4).
Simvastatin
Simvastatin ist ein Substrat von Cytochrom P450 3A4. Potente Inhibitoren von Cytochrom P450 3A4 erhöhen das Risiko für eine Myopathie und Rhabdomyolyse durch die Erhöhung der Konzentration der inhibitorischen Aktivität der HMG-CoA-Reduktase im Plasma während der Therapie mit Simvastatin.
Zu diesen Inhibitoren zählen Itraconazol, Ketoconazol, Posaconazol, Voriconazol, Erythromycin, Clarithromycin, Telithromycin, HIV-Protease-Inhibitoren (z. B. Nelfinavir), Boceprevir, Telaprevir, Nefazodon und Arzneimittel, die Cobicistat enthalten. Die gleichzeitige Anwendung von Itraconazol führte zu einer mehr als zehnfachen Erhöhung der Exposition mit Simvastatinsäure (aktiver
Betahydroxysäure-Metabolit). Telithromycin führte zu einer elffachen Erhöhung der Exposition mit Simvastatinsäure.
Eine gleichzeitige Anwendung mit Itraconazol, Ketoconazol, Posaconazol, Voriconazol, HIV-Protease-Inhibitoren (z. B. Nelfinavir), Boceprevir, Telaprevir, Erythromycin, Clarithromycin, Telithromycin, Nefazodon und Arzneimittel, die Cobicistat enthalten, sowie Gemfibrozil, Ciclosporin und Danazol ist kontraindiziert (siehe Abschnitt 4.3). Falls eine Behandlung mit potenten CYP3A4-Inhibitoren (Substanzen, welche die AUC mindestens um ca. das 5-Fache erhöhen) unabdingbar ist, muss die Therapie mit GOLTOR während der Behandlungsdauer unterbrochen werden (und die Anwendung eines alternativen Statins in Erwägung gezogen werden). Vorsicht ist angebracht, wenn GOLTOR mit bestimmten anderen weniger potenten CYP3A4-Inhibitoren kombiniert wird: Fluconazol, Verapamil oder Diltiazem (siehe Abschnitte 4.2 und 4.4).
Fluconazol: Bei der kombinierten Anwendung von Simvastatin und Fluconazol wurden seltene Fälle von Rhabdomyolyse berichtet (siehe Abschnitt 4.4).
Ciclosporin: Das Risiko für eine Myopathie/Rhabdomyolyse wird durch die gleichzeitige Anwendung von Ciclosporin mit GOLTOR erhöht. Daher ist die gleichzeitige Gabe von Ciclosporin kontraindiziert (siehe Abschnitte 4.3 und 4.4). Obwohl der Mechanismus noch nicht vollständig geklärt ist, wurde gezeigt, dass Ciclosporin die AUC von HMG-CoA-Reduktase-Inhibitoren vergrößert. Die Vergrößerung der AUC der Simvastatinsäure ist vermutlich teilweise auf eine Hemmung von CYP3A4 und/oder von OATP1B1 zurückzuführen.
Danazol: Das Risiko für eine Myopathie/Rhabdomyolyse ist durch die gleichzeitige Anwendung von Danazol mit GOLTOR erhöht. Daher ist die gleichzeitige Gabe von Danazol kontraindiziert (siehe Abschnitte 4.3 und 4.4).
Gemfibrozil: Gemfibrozil erhöht die AUC der Simvastatinsäure um das 1,9-Fache, möglicherweise aufgrund einer Hemmung des Glukuronidierungsweges und/oder von OATP1B1 (siehe Abschnitte 4.3 und 4.4). Die gleichzeitige Gabe von Gemfibrozil ist kontraindiziert.
Fusidinsäure: Das Risiko einer Myopathie einschließlich Rhabdomyolyse kann bei gleichzeitiger systemischer Gabe von Fusidinsäure und Statinen erhöht sein. Die gleichzeitige Gabe dieser Kombination kann zu erhöhten Plasmaspiegeln beider Substanzen führen. Der dieser Wechselwirkung zugrundeliegende Mechanismus (ob pharmakodynamisch oder pharmakokinetisch oder beiderseits begründet) ist derzeit noch nicht geklärt. Es wurde über das Auftreten von Rhabdomyolyse (einschließlich einiger mit Todesfolge) bei Patienten berichtet, welche diese Kombination erhielten. Sofern die Behandlung mit Fusidinsäure notwendig ist, ist GOLTOR während der gesamten Behandlungsdauer mit Fusidinsäure abzusetzen (siehe Abschnitt 4.4).
Amiodaron: Das Risiko für Myopathie und Rhabdomyolyse ist bei gleichzeitiger Therapie mit Amiodaron und Simvastatin erhöht (siehe Abschnitt 4.4). In einer klinischen Studie wurde bei 6 % der Patienten, die 80 mg Simvastatin und Amiodaron einnahmen, über eine Myopathie berichtet. Die Dosis von GOLTOR sollte daher 10 mg/20 mg pro Tag bei Kombination mit Amiodaron nicht überschreiten.
Kalziumkanalblocker
• Verapamil
Das Risiko einer Myopathie und Rhabdomyolyse ist bei gleichzeitiger Anwendung von Verapamil und 40 mg oder 80 mg Simvastatin erhöht (siehe Abschnitt 4.4). In einer pharmakokinetischen Studie führte eine gleichzeitige Anwendung von Simvastatin mit Verapamil zu einer 2,3fachen Erhöhung der Exposition mit Simvastatinsäure, was vermutlich teilweise auf eine CYP3A4-Hemmung zurückzuführen ist. Die Dosis von GOLTOR sollte daher 10 mg/20 mg pro Tag bei Kombination mit Verapamil nicht überschreiten.
• Diltiazem
Das Risiko einer Myopathie und Rhabdomyolyse ist bei gleichzeitiger Anwendung von Diltiazem und 80 mg Simvastatin erhöht (siehe Abschnitt 4.4). In einer pharmakokinetischen Studie führte die gleichzeitige Anwendung von Simvastatin mit Diltiazem zu einer 2,7fachen Erhöhung der Exposition mit Simvastatinsäure, was vermutlich auf eine CYP3A4-Hemmung zurückzuführen ist. Die Dosis von GOLTOR sollte daher 10 mg/20 mg pro Tag bei Kombination mit Diltiazem nicht überschreiten.
• Amlodipin
Für Patienten unter Amlodipin, die gleichzeitig Simvastatin erhalten, besteht ein erhöhtes Myopathierisiko. In einer pharmakokinetischen Studie führte eine gleichzeitige Anwendung mit Amlodipin zu einer ca. 1,6fachen Erhöhung der Exposition mit der Simvastatinsäure. Die Dosis von GOLTOR sollte daher 10 mg/20 mg pro Tag bei Kombination mit Amlodipin nicht überschreiten.
Lomitapid: Das Risiko für Myopathie und Rhabdomyolyse kann durch die gemeinsame Anwendung von Lomitapid und Simvastatin erhöht sein (siehe Abschnitte 4.3 und 4.4). Deshalb darf bei Patienten mit homozygoter familiärer Hypercholesterinämie (HoFH) unter Lomitapid eine Dosis von GOLTOR 10 mg/40 mg nicht überschritten werden.
Moderate CYP3A4-Inhibitoren: Patienten, die gleichzeitig mit GOLTOR (vor allem hohe GOLTOR Dosierungen) andere Arzneimittel einnehmen, die bei therapeutischer Dosierung moderate CYP3A4-Inhibitoren sind, könnten ein erhöhtes Myopathierisiko haben (siehe Abschnitt 4.4).
Inhibitoren des OATP1B1-Transporterproteins: Simvastatinsäure ist ein Substrat für das OATP1B1-Transporterprotein. Die gemeinsame Anwendung von Arzneimitteln, die Inhibitoren des OATP1B1-Transporterproteins sind, kann zu erhöhten Plasmaspiegeln von Simvastatinsäure führen, und damit zu einer Erhöhung des Myopathierisikos (siehe Abschnitte 4.3 und 4.4).
Grapefruitsaft: Grapefruitsaft hemmt Cytochrom P450 3A4. Der Genuss großer Mengen von Grapefruitsaft (über 1 Liter pro Tag) bei gleichzeitiger Anwendung von Simvastatin führte zu einer 7fachen Erhöhung der Exposition mit Simvastatinsäure. Der Genuss von 240 ml Grapefruitsaft am Morgen und die Einnahme von Simvastatin am Abend führte ebenso zu einer 1,9fachen Erhöhung.
Der Genuss von Grapefruitsaft sollte deshalb während der Therapie mit GOLTOR vermieden werden.
Colchicin: Bei Patienten mit eingeschränkter Nierenfunktion wurde über Myopathie und Rhabdomyolyse unter gleichzeitiger Anwendung von Colchicin und Simvastatin berichtet. Eine engmaschige klinische Überwachung betroffener Patienten, die diese Kombination einnehmen, wird angeraten.
Rifampicin: Da Rifampicin ein starker CYP3A4-Induktor ist, kann es bei Patienten unter Dauertherapie mit Rifampicin (z. B. bei Behandlung einer Tuberkulose) zu einer Verringerung der Wirksamkeit von Simvastatin kommen. In einer pharmakokinetischen Studie mit gesunden Probanden war unter gleichzeitiger Anwendung von Rifampicin die Fläche unter der Plasmakonzentrationskurve (AUC) für Simvastatinsäure um 93 % erniedrigt.
Niacin: Es wurden Fälle von Myopathie/Rhabdomyolyse unter Simvastatin in Kombination mit lipidsenkenden Dosen (> 1 g/Tag) von Niacin beobachtet (siehe Abschnitt 4.4).
Wirkungen von GOLTOR auf die Pharmakokinetik anderer Arzneimittel
Ezetimib
In präklinischen Studien wurde gezeigt, dass Ezetimib die Enzyme des Cytochrom-P450-Metabolismus nicht induziert. Es wurden keine klinisch bedeutenden pharmakokinetischen Wechselwirkungen zwischen Ezetimib und Arzneimitteln beobachtet, die bekanntermaßen über Cytochrom P450 1A2, 2D6, 2C8, 2C9 und 3A4 oder N-Acetyltransferase metabolisiert werden.
Antikoagulanzien: In einer Studie an 12 gesunden erwachsenen Männern hatte die gleichzeitige Anwendung von Ezetimib (10 mg einmal täglich) keine signifikante Wirkung auf die Bioverfügbarkeit von Warfarin und auf die Prothrombinzeit. Nach Markteinführung wurde jedoch über Erhöhungen der „International Normalized Ratio“ (INR) bei Patienten unter Warfarin- oder Fluindion-Therapie berichtet, die zusätzlich Ezetimib erhielten. Bei Zugabe von GOLTOR zu Warfarin, einem anderen Cumarin-Antikoagulans oder Fluindion ist die „International Normalized Ratio“ (INR) entsprechend zu überwachen (siehe Abschnitt 4.4).
Simvastatin
Simvastatin übt keine inhibitorische Wirkung auf Cytochrom P450 3A4 aus. Daher wird auch keine Wirkung von Simvastatin auf die Plasmakonzentrationen von über CYP3A4 metabolisierten Substanzen erwartet.
Orale Antikoagulanzien: In zwei klinischen Studien, von denen die eine mit gesunden Probanden, die andere mit Patienten mit Hypercholesterinämie durchgeführt wurde, führte Simvastatin 20-40 mg/Tag zu einer moderaten Wirkungsverstärkung von Antikoagulanzien vom Typ der Cumarin-Derivate. Die Prothrombinzeit, angegeben in der „International Normalized Ratio“ (INR), erhöhte sich bei den Probanden von 1,7 auf 1,8 und bei den Patienten von 2,6 auf 3,4. Es wurden sehr seltene Fälle von Erhöhungen der INR berichtet. Daher sollte bei Patienten, die Cumarin-Derivate einnehmen, die Prothrombinzeit vor Beginn einer Therapie mit GOLTOR und danach zu Beginn der Therapie in häufigen Abständen bestimmt werden, um signifikante Veränderungen der Prothrombinzeit zu verhindern. Nach Stabilisierung der Werte wird die Bestimmung der Prothrombinzeit anschließend in den Zeitabständen empfohlen, wie sie für Patienten unter der Therapie mit Cumarin-Derivaten üblich sind. Wird die Dosis von GOLTOR geändert oder GOLTOR abgesetzt, sollte dieselbe Vorgehensweise eingehalten werden. Die Therapie mit Simvastatin wurde nicht mit Blutungen oder Veränderungen der Prothrombinzeit bei Patienten, die keine Antikoagulanzien einnahmen, in Zusammenhang gebracht.
Kinder und Jugendliche
Studien zur Erfassung von Wechselwirkungen wurden nur bei Erwachsenen durchgeführt.
4.6 Fertilität, Schwangerschaft und Stillzeit
Schwangerschaft
Da Atherosklerose eine chronische Erkrankung ist, sollte eine Unterbrechung der lipidsenkenden Therapie während einer Schwangerschaft normalerweise kaum Einfluss auf das mit einer primären Hypercholesterinämie verbundene Langzeitrisiko haben.
GOLTOR
GOLTOR ist während der Schwangerschaft kontraindiziert. Zur Anwendung von GOLTOR während einer Schwangerschaft liegen keine klinischen Daten vor. Tierstudien zur Kombinationstherapie haben Reproduktionstoxizität gezeigt (siehe Abschnitt 5.3).
Simvastatin
Die Sicherheit von Simvastatin bei schwangeren Frauen wurde nicht untersucht. Mit Simvastatin wurden keine kontrollierten klinischen Studien mit schwangeren Frauen durchgeführt. Es liegen seltene Berichte über kongenitale Anomalien nach intrauteriner Exposition mit HMG-CoA-Reduktase-Inhibitoren vor. Eine Analyse bisheriger Erfahrungen mit ca. 200 Frauen, die versehentlich Simvastatin oder einen strukturverwandten HMG-CoA-Reduktase-Inhibitor im ersten Trimenon der Schwangerschaft eingenommen hatten, zeigte kein gegenüber der Gesamtpopulation erhöhtes Risiko für kongenitale Anomalien. Diese Fallzahl war statistisch ausreichend, um eine Risikoerhöhung um das 2,5-Fache oder mehr im Vergleich zu der für eine Gesamtpopulation erwarteten Häufigkeit ausschließen zu können.
Obwohl es keine Anzeichen dafür gibt, dass die Inzidenz kongenitaler Anomalien bei Kindern, deren Mütter Simvastatin oder einen anderen eng verwandten HMG-CoA-Reduktase-Inhibitor eingenommen hatten, von der in der Gesamtpopulation beobachteten abweicht, kann eine Behandlung der Mutter mit Simvastatin beim Fetus die Spiegel der Mevalonsäure senken, welche als Vorstufe der Cholesterinsynthese eine Rolle spielt. GOLTOR darf daher nicht von Frauen eingenommen werden, die schwanger sind, eine Schwangerschaft planen oder vermuten schwanger zu sein. Die Behandlung mit GOLTOR muss unterbrochen werden, bis die Schwangerschaft beendet oder definitiv ausgeschlossen ist (siehe Abschnitt 4.3).
Ezetimib
Es liegen keine klinischen Daten zur Anwendung von Ezetimib während einer Schwangerschaft vor. Stillzeit
GOLTOR ist während der Stillzeit kontraindiziert. Studien an Ratten haben gezeigt, dass Ezetimib in die Muttermilch übergeht. Es ist nicht bekannt, ob die Wirkstoffe von GOLTOR in die menschliche Muttermilch übergehen (siehe Abschnitt 4.3).
Fertilität
Ezetimib
Es liegen keine Daten aus klinischen Studien zu den Auswirkungen von Ezetimib auf die menschliche Fertilität vor. Ezetimib hatte keine Auswirkungen auf die Fertilität von weiblichen oder männlichen Ratten (siehe Abschnitt 5.3).
Simvastatin
Es liegen keine Daten aus klinischen Studien zu den Auswirkungen von Simvastatin auf die menschliche Fertilität vor. Simvastatin zeigte keine Auswirkungen auf die Fertilität von männlichen und weiblichen Ratten (siehe Abschnitt 5.3).
4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Es wurden keine Studien zu den Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen durchgeführt. Bei der Teilnahme am Straßenverkehr oder dem Bedienen von Maschinen ist jedoch zu berücksichtigen, dass über Schwindel berichtet wurde.
4.8 Nebenwirkungen
Tabellarische Übersicht der Nebenwirkungen (Klinische Studien)
Die Sicherheit von GOLTOR (oder der Koadministration von Ezetimib und Simvastatin, äquivalent mit der Einnahme von GOLTOR) wurde bei ca. 12.000 Patienten in klinischen Studien untersucht.
Bei der Bewertung von Nebenwirkungen werden folgende Häufigkeiten zugrunde gelegt:
Sehr häufig (> 1/10), häufig (> 1/100, < 1/10), gelegentlich (> 1/1.000, < 1/100), selten (> 1/10.000,
< 1/1.000), sehr selten (< 1/10.000) einschließlich gemeldeter Einzelfälle
Folgende Nebenwirkungen wurden bei mit GOLTOR behandelten Patienten (n = 2.404) häufiger als unter Plazebo (n = 1.340) beobachtet (siehe Tabelle 2).
|
Systemorganklasse |
Nebenwirkung |
Häufigkeit |
|
Untersuchungen |
Erhöhungen von ALT und/oder AST, Erhöhungen der CK im Serum |
Häufig |
|
Erhöhte Bilirubin-Werte, erhöhte Blutharnsäure, erhöhte y-Glutamyltranspeptidase, erhöhte INR, Protein im Urin, Gewichtsabnahme |
Gelegentlich | |
|
Erkrankungen des Nervensystems |
Schwindel, Kopfschmerzen |
Gelegentlich |
|
Erkrankungen des Gastrointe stinaltrakts |
Abdominalschmerzen, Abdominalbe schwerden, Oberbauchschmerzen, Dyspepsie, Flatulenz, Übelkeit, Erbrechen |
Gelegentlich |
|
Erkrankungen der Haut und des Unterhautzellgewebes |
Pruritus, Hautausschlag |
Gelegentlich |
|
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen |
Arthralgie, Muskelspasmen, Muskelschwäche, muskuloskelettale Beschwerden, Nackenschmerzen, Schmerzen in den Gliedmaßen |
Gelegentlich |
|
Allgemeine Erkrankungen und Beschwerden am V erabreichungsort |
Asthenie, Abgeschlagenheit, Unwohlsein, periphere Ödeme |
Gelegentlich |
|
Psychiatrische Erkrankungen |
Schlafstörungen |
Gelegentlich |
Folgende Nebenwirkungen wurden bei mit GOLTOR behandelten Patienten (n = 9.595) häufiger als unter Statinen allein (n = 8.883) beobachtet (siehe Tabelle 3).
|
Systemorganklasse |
Nebenwirkungen |
Häufigkeit |
|
Untersuchungen |
Erhöhungen von ALT und/oder AST |
Häufig |
|
Erhöhte Bilirubin-Werte, erhöhte CK im Blut, erhöhte y-Glutamyltranspeptidase |
Gelegentlich | |
|
Erkrankungen des Nervensystems |
Kopfschmerzen, Parästhesien |
Gelegentlich |
|
Erkrankungen des Gastrointe stinaltrakts |
Geblähtes Abdomen, Diarrhö; trockener Mund, Dyspepsie, Flatulenz, gastroösophagealer Reflux, Erbrechen |
Gelegentlich |
|
Erkrankungen der Haut und des Unterhautzellgewebes |
Pruritus, Hautausschlag, Urtikaria |
Gelegentlich |
|
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen |
Myalgie |
Häufig |
|
Arthralgie, Rückenschmerzen, Muskelspasmen, Muskelschwäche, muskuloskelettale Schmerzen, Schmerzen in den Gliedmaßen |
Gelegentlich | |
|
Allgemeine Erkrankungen und Beschwerden am V erabreichungsort |
Asthenie, Schmerzen im Brustkorb, Fatigue-Syndrom, periphere Ödeme |
Gelegentlich |
|
Psychiatrische Erkrankungen |
Schlaflosigkeit |
Gelegentlich |
Kinder und Jugendliche
In einer Studie mit heranwachsenden Patienten (10 bis 17 Jahre) mit heterozygoter familiärer Hypercholesterinämie (n = 248) wurden Erhöhungen von ALT und/oder AST (> dem Dreifachen des oberen Normwertes in Folge) bei 3 % (4 Patienten) der Patienten unter Ezetimib/Simvastatin beobachtet, im Vergleich zu 2 % (2 Patienten) unter Simvastatin-Monotherapie; für CPK-Erhöhungen (> dem Zehnfachen des oberen Normwertes) lagen diese Werte bei 2 % (2 Patienten) bzw. bei 0 %. Es wurden keine Fälle von Myopathie berichtet.
Diese Studie war nicht zum Vergleich seltener Nebenwirkungen geeignet.
Patienten mit koronarer Herzkrankheit und akutem Koronarsyndrom in der Vorgeschichte In der IMPROVE-IT-Studie (siehe Abschnitt 5.1), in der 18.144 Patienten entweder mit GOLTOR 10 mg/40 mg (n = 9.067; 6 % der Patienten wurden auf GOLTOR 10 mg/80 mg hochtitriert) oder Simvastatin 40 mg (n = 9.077; 27 % der Patienten wurden auf Simvastatin 80 mg hochtitriert) behandelt wurden, zeigten sich innerhalb der medianen Nachbeobachtung von 6,0 Jahren ähnliche Sicherheitsprofile bei beiden Behandlungsgruppen. Die Behandlungsabbruchrate aufgrund von Nebenwirkungen betrug 10,6 % bei Patienten unter GOLTOR und 10,1 % bei Patienten unter Simvastatin. Die Inzidenz einer Myopathie betrug 0,2 % in der GOLTOR-Gruppe und 0,1 % in der Simvastatin-Monotherapie-Gruppe. Myopathie war definiert als Muskelschwäche oder Muskelschmerzen ungeklärter Ursache mit einer Erhöhung des Serumkreatinins (CK) um das > 10Fache des oberen Normwertes [ULN] oder zwei aufeinanderfolgenden Erhöhungen des Serumkreatinins (CK) um das > 5 - < 10-Fache des oberen Normwertes [ULN]. Die Inzidenz einer Rhabdomyolyse betrug 0,1 % in der GOLTOR-Gruppe und 0,2 % in der Simvastatin-Monotherapie-Gruppe. Rhabdomyolyse war definiert als Muskelschwäche oder Muskelschmerzen ungeklärter Ursache mit einer Erhöhung des Serumkreatinins (CK) um das > 10-Fache des oberen Normwertes [ULN] mit Nachweis einer Nierenschädigung oder zwei aufeinanderfolgenden Erhöhungen des Serumkreatinins (CK) um das > 5 - < 10-Fache des oberen Normwertes [ULN] mit Nachweis einer Nierenschädigung oder mit einem Serumkreatinin (CK) von > 10.000 IU/l ohne Nachweis einer Nierenschädigung. Die Inzidenz einer konsekutiven Erhöhung der Transaminasenwerte (> dem Dreifachen des oberen Normwertes [ULN]) betrug 2,5 % in der GOLTOR-Gruppe und 2,3 % in der
Simvastatin-Monotherapie-Gruppe (siehe Abschnitt 4.4). Nebenwirkungen in Verbindung mit der Gallenblase wurden bei 3,1 % der Patienten unter GOLTOR im Vergleich zu 3,5 % der Patienten unter Simvastatin berichtet. Die Inzidenz stationärer Einweisungen aufgrund einer Cholezystektomie betrug 1,5 % bei beiden Behandlungsgruppen. Krebserkrankungen (definiert als jegliche neu diagnostizierte maligne Erkrankung) wurden im Verlauf der Studie bei 9,4 % beziehungsweise 9,5 % der Patienten diagnostiziert.
Patienten mit chronischer Nierenerkrankung
In der SHARP-Studie („Study of Heart and Renal Protection“) (siehe Abschnitt 5.1), in der mehr als
9.000 Patienten mit GOLTOR 10 mg/20 mg einmal täglich (n = 4.650) oder Plazebo (n = 4.620) behandelt wurden, ergaben sich bei einer medianen Verlaufsbeobachtung von 4,9 Jahren vergleichbare Sicherheitsprofile zwischen beiden Patientengruppen. In dieser Studie wurden lediglich schwerwiegende Nebenwirkungen und Behandlungsabbrüche als Folge jeglicher Nebenwirkungen erfasst. Die Abbruchraten aufgrund von Nebenwirkungen waren vergleichbar (10,4 % der Patienten unter GOLTOR und 9,8 % der Patienten unter Plazebo). Die Inzidenz für Myopathie/Rhabdomyolyse betrug bei den mit GOLTOR behandelten Patienten 0,2 % und 0,1 % in der Plazebo-Gruppe. Eine konsekutive Erhöhung der Transaminasenwerte (> dem Dreifachen des oberen Normwertes [ULN]) wurde bei 0,7 % der Patienten unter GOLTOR und bei 0,6 % der Patienten in der Plazebo-Gruppe festgestellt (siehe Abschnitt 4.4). Im Rahmen dieser Studie wurde keine statistisch signifikante Erhöhung der Inzidenz von vorab definierten Nebenwirkungen festgestellt, einschließlich Krebserkrankungen (9,4 % unter GOLTOR, 9,5 % unter Plazebo), Hepatitis, Cholezystektomie oder Komplikationen mit Gallensteinen oder Pankreatitis.
Laborwerte
In Koadministrationsstudien betrug die Inzidenz klinisch bedeutender Erhöhungen der Serum-Transaminasen (ALT und/oder AST > dem Dreifachen des oberen Normwertes in Folge) 1,7 % unter GOLTOR. Diese Erhöhungen waren im Allgemeinen asymptomatisch, standen nicht im Zusammenhang mit einer Cholestase und kehrten nach Absetzen der Therapie oder bei Fortsetzung der Behandlung auf den Ausgangswert zurück (siehe Abschnitt 4.4).
Klinisch bedeutende Erhöhungen der CK (> dem Zehnfachen des oberen Normwertes) wurden bei 0,2 % der mit GOLTOR behandelten Patienten beobachtet.
Erfahrungen nach Markteinführung
Die folgenden Nebenwirkungen wurden zusätzlich nach Markteinführung unter GOLTOR bzw. in klinischen Studien oder nach Markteinführung unter einem der einzelnen Bestandteile berichtet.
Erkrankungen des Blutes und des Lymphsystems: Thrombozytopenie, Anämie
Erkrankungen des Nervensystems: periphere Neuropathie, Beeinträchtigung des Erinnerungsvermögens
Erkrankungen der Atemwege, des Brustraums und Mediastinums: Husten, Dyspnoe, interstitielle Lungenkrankheit (siehe Abschnitt 4.4)
Erkrankungen des Gastrointestinaltrakts: Obstipation, Pankreatitis, Gastritis
Erkrankungen der Haut und des Unterhautzellgewebes: Alopezie, Erythema multiforme, Überempfindlichkeitsreaktionen einschließlich Hautausschlag, Urtikaria, Anaphylaxie, Angioödem
Skelettmuskulatur- und Bindegewebserkrankungen: Muskelkrämpfe, Myopathie* (einschl. Myositis), Rhabdomyolyse mit oder ohne akutem Nierenversagen (siehe Abschnitt 4.4), Tendinopathie, gelegentlich bis hin zur Sehnenruptur, immunvermittelte nekrotisierende Myopathie (Häufigkeit nicht bekannt)** * Myopathie trat in einer klinischen Studie häufig bei Patienten unter 80 mg Simvastatin pro Tag auf (1,0 %), im Vergleich zu Patienten unter 20 mg Simvastatin pro Tag (0,02 %) (siehe Abschnitte 4.4 und 4.5).
** In sehr seltenen Fällen wurde während oder nach der Behandlung mit einigen Statinen über eine autoimmunvermittelte nekrotisierende Myopathie (immune-mediated necrotizing myopathy; IMNM) berichtet. Die klinischen Charakteristika einer IMNM sind persistierende proximale Muskelschwäche und erhöhte Serum-Kreatinkinase-Werte, die trotz Absetzen der Behandlung mit Statinen fortbestehen. Muskelbiopsien zeigen eine nekrotisierende Myopathie ohne signifikante Entzündungen. Eine Besserung zeigt sich unter Anwendung von Immunsuppressiva (siehe Abschnitt 4.4).
Stoffwechsel- und Ernährungsstörungen: verminderter Appetit
Gefäßerkrankungen: Hitzewallungen, Hypertonie
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort: Schmerzen
Leber- und Gallenerkrankungen: Hepatitis/Ikterus, Leberversagen mit teils tödlichem Ausgang, Cholelithiasis, Cholezystitis
Erkrankungen der Geschlechtsorgane und der Brustdrüse: erektile Dysfunktion Psychiatrische Erkrankungen: Depression, Schlaflosigkeit
Selten wurde über ein offensichtliches Hypersensitivitätssyndrom berichtet, das mit einigen der folgenden Symptome einherging: angioneurotisches Ödem, Lupus-ähnliches Syndrom, Polymyalgia rheumatica, Dermatomyositis, Vaskulitis, Thrombozytopenie, Eosinophilie, Beschleunigung der Blutsenkungsgeschwindigkeit, Arthritis und Arthralgie, Urtikaria, Photosensitivität, Fieber, Hitzewallung (Flushing), Dyspnoe und allgemeines Krankheitsgefühl.
Laborwerte: Erhöhungen der alkalischen Phosphatase, Abweichungen in Leberfunktionstests
Erhöhungen von HbA1c und Nüchternglucosespiegel wurden im Zusammenhang mit Statinen, einschließlich Simvastatin berichtet.
Selten wurde nach Markteinführung im Zusammenhang mit der Einnahme von Statinen einschließlich Simvastatin über kognitive Beeinträchtigungen (z. B. Gedächtnisverlust, Vergesslichkeit, Amnesie, Gedächtnisstörungen, Verwirrung) berichtet. Diese sind im Allgemeinen nicht schwerwiegend und nach Absetzen der Statine reversibel, mit unterschiedlichen Zeitspannen bis zum Auftreten (von 1 Tag bis zu Jahren) und Abklingen (3 Wochen im Median) der Symptome.
Die folgenden Nebenwirkungen wurden bei einigen Statinen berichtet:
• Schlafstörungen einschließlich Alpträume
• Störungen der Sexualfunktion
• Diabetes mellitus: Die Häufigkeit ist abhängig von dem Vorhandensein oder dem Fehlen von Risikofaktoren (Nüchternblutzucker > 5,6 mmol/l, BMI > 30 kg/m2, erhöhte Triglyzeridwerte, bestehende Hypertonie).
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Arzneimittel und Medizinprodukte, Abt. Pharmakovigilanz, Kurt-Georg-Kiesinger-Allee 3, D-53175 Bonn, Website: www.bfarm.de anzuzeigen.
4.9 Überdosierung
GOLTOR
Im Fall einer Überdosierung sollten symptomatische und unterstützende Maßnahmen ergriffen werden. Eine Koadministration von Ezetimib (1.000 mg/kg) und Simvastatin (1.000 mg/kg) wurde von Mäusen und Ratten in Studien zur oralen akuten Toxizität gut vertragen. Bei diesen Tieren wurden keine klinischen Anzeichen einer toxischen Wirkung beobachtet. Die geschätzte orale LD50 lag für beide Spezies über 1.000 mg/kg Ezetimib sowie über 1.000 mg/kg Simvastatin.
Ezetimib
In klinischen Studien wurde die Gabe von 50 mg Ezetimib/Tag bei 15 Probanden bis zu 14 Tage lang wie auch die Gabe von 40 mg/Tag an 18 Patienten mit primärer Hypercholesterinämie bis zu 56 Tage lang im Allgemeinen gut vertragen. Einige Fälle von Überdosierung wurden berichtet, die meist nicht von unerwünschten Ereignissen begleitet waren. Die unerwünschten Ereignisse, die dabei berichtet wurden, waren nicht schwerwiegend. Bei Tieren wurden nach oral verabreichten Einzeldosen von
5.000 mg Ezetimib/kg an Ratten und Mäusen sowie von 3.000 mg Ezetimib/kg an Hunden keine toxischen Effekte beobachtet.
Simvastatin
Einige Fälle von Überdosierung wurden berichtet. Die höchste eingenommene Dosis betrug 3,6 g Simvastatin. Bei keinem der Patienten kam es zu Folgeerscheinungen.
5. PHARMAKOLOGISCHE EIGENSCHAFTEN
5.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: HMG-CoA-Reduktase-Hemmer in Kombination mit anderen Lipidsenkern, ATC-Code: C10B A02
GOLTOR (Ezetimib/Simvastatin) ist ein lipidsenkendes Präparat, das selektiv die intestinale Resorption von Cholesterin und verwandten Phytosterinen hemmt und die endogene Cholesterinsynthese reduziert.
Wirkmechanismus
GOLTOR
Das Cholesterin im Plasma stammt aus der intestinalen Resorption und der endogenen Synthese. GOLTOR enthält Ezetimib und Simvastatin, zwei lipidsenkende Stoffe mit komplementären Wirkmechanismen. GOLTOR senkt erhöhte Werte von Gesamtcholesterin, LDL-Cholesterin, Apolipoprotein B, Triglyzeriden, non-HDL-Cholesterin und erhöht den HDL-Cholesterinwert durch die duale Hemmung der Cholesterinresorption und -synthese.
Ezetimib
Ezetimib hemmt die intestinale Cholesterinresorption. Ezetimib ist nach oraler Einnahme wirksam; seine Wirkungsweise unterscheidet sich von der anderer Klassen von cholesterinsenkenden Stoffen (z. B. Statine, Anionenaustauscher [Harze], Fibrinsäurederivate und Phytosterine). Das molekulare Ziel von Ezetimib ist der Steroltransporter, das Niemann-Pick-C1 Like 1 (NPC1L1) Protein, der für die intestinale Aufnahme von Cholesterin und Phytosterinen verantwortlich ist.
Ezetimib lagert sich am Bürstensaum des Dünndarms an und hemmt die Cholesterinresorption, was zu einem verminderten Transport von Cholesterin aus dem Darm in die Leber führt. Statine reduzieren die Cholesterinsynthese in der Leber, und gemeinsam führen diese unterschiedlichen Wirkungsmechanismen zu einer komplementären Cholesterinsenkung. In einer zweiwöchigen klinischen Studie an 18 Patienten mit Hypercholesterinämie hemmte Ezetimib im Vergleich zu Plazebo die intestinale Cholesterinresorption um ca. 54 %.
Eine Reihe von präklinischen Studien wurde durchgeführt, um die Selektivität von Ezetimib für die Hemmung der Cholesterinresorption zu bestimmen. Ezetimib hemmte die Resorption von radioaktiv markiertem [14C]Cholesterin ohne Wirkung auf die Resorption von Triglyzeriden, Fettsäuren, Gallensäuren, Progesteron, Ethinylestradiol oder der fettlöslichen Vitamine A und D.
Simvastatin
Nach oraler Aufnahme wird Simvastatin, ein inaktives Lacton, in der Leber zur entsprechenden aktiven Betahydroxysäure hydrolysiert. Diese ist ein starker Inhibitor der 3-Hydroxy-3-methylglutaryl-Coenzym-A(HMG-CoA)-Reduktase. Dieses Enzym katalysiert die Umwandlung von HMG-CoA zu Mevalonat, einen frühen und geschwindigkeitsbestimmenden Schritt in der Biosynthese des Cholesterins.
Simvastatin senkt erwiesenermaßen normale und erhöhte Werte von LDL-Cholesterin. LDL entsteht aus VLDL und wird überwiegend über spezifische LDL-Rezeptoren abgebaut. Der LDL-senkende Wirkmechanismus von Simvastatin beruht wahrscheinlich sowohl auf der Abnahme der Konzentration von VLDL-Cholesterin als auch auf einer Induktion von LDL-Rezeptoren und somit auf einer verminderten Produktion als auch auf einem verstärkten Abbau von LDL-Cholesterin. Die Konzentration von Apolipoprotein B nimmt bei der Behandlung mit Simvastatin ebenfalls deutlich ab. Simvastatin bewirkt außerdem einen moderaten Anstieg des HDL-Cholesterins sowie eine Abnahme der Triglyzeride im Plasma. Insgesamt resultiert aus diesen Veränderungen eine Abnahme der Verhältnisse von Gesamt- zu HDL-Cholesterin und LDL- zu HDL-Cholesterin.
Klinische Wirksamkeit und Sicherheit
In kontrollierten klinischen Studien führte GOLTOR zu einer signifikanten Reduktion der Werte von Gesamtcholesterin, LDL-Cholesterin, Apolipoprotein B, Triglyzeriden und non-HDL-Cholesterin und einer Erhöhung von HDL-Cholesterin bei Patienten mit Hypercholesterinämie.
Prävention kardiovaskulärer Ereignisse
Für GOLTOR konnte bei Patienten mit koronarer Herzkrankheit und akutem Koronarsyndrom in der Vorgeschichte eine Verringerung von schweren (major) kardiovaskulären Ereignissen nachgewiesen werden.
Im Rahmen der IMPROVE-IT-Studie (IMProved Reduction of Outcomes: Vytorin Efficacy International Trial), einer multizentrischen, randomisierten, doppelblinden, aktiv-kontrollierten Studie wurden 18.144 Patienten untersucht, die innerhalb von 10 Tagen nach stationärer Einweisung aufgrund eines akuten Koronarsyndroms (entweder akuter Myokardinfarkt [MI] oder instabile Angina pectoris [UA]) in die Studie eingeschlossen wurden. Bei Vorstellung mit akutem Koronarsyndrom hatten die Patienten, die nicht mit einer lipidsenkenden Therapie vorbehandelt waren LDL-Cholesterinwerte von < 125 mg/dl (< 3,2 mmol) und Patienten, die bereits mit einer lipidsenkenden Therapie vorbehandelt waren < 100 mg/dl (< 2,6 mmol). Alle Patienten erhielten randomisiert 1:1 entweder Ezetimib/Simvastatin 10 mg/40 mg (n = 9.067) oder Simvastatin 40 mg (n = 9.077) und wurden im Median über 6,0 Jahre nachbeobachtet.
Die Patienten waren im Mittel 63,6 Jahre alt, 76 % waren Männer, 84 % waren kaukasischer Herkunft und 27 % waren Diabetiker. Der durchschnittliche LDL-Cholesterinwert zum Zeitpunkt des Studieneinschlussereignisses lag bei den Patienten unter lipidsenkender Vortherapie (n = 6.390) bei 80 mg/dl (2,1 mmol/l) und bei den Patienten ohne lipidsenkende Vortherapie (n = 11.594) bei 101 mg/dl (2,6 mmol/l). Vor der stationären Aufnahme aufgrund von akutem Koronarsyndrom (Studieneinschlussereignis) erhielten 34 % der Patienten eine Vortherapie mit einem Statin. Zum Untersuchungszeitpunkt nach einem Jahr lag der durchschnittliche LDL-Cholesterinwert unter fortlaufender Behandlung bei den Patienten in der GOLTOR-Gruppe bei 53,2 mg/dl (1,4 mmol/l) und in der Simvastatin-Monotherapie-Gruppe bei 69,9 mg/dl (1,8 mmol/l). Bei den Patienten unter fortlaufender Studienmedikation wurden grundsätzlich die Lipidwerte erhoben.
Der primäre Endpunkt war eine Kombination der Ereignisse kardiovaskulärer Tod, schwere (major) koronare Ereignisse (MCE; definiert als nicht-tödlicher Myokardinfarkt, nachgewiesene instabile Angina pectoris mit erforderlicher stationärer Einweisung oder jegliche, mindestens 30 Tage nach
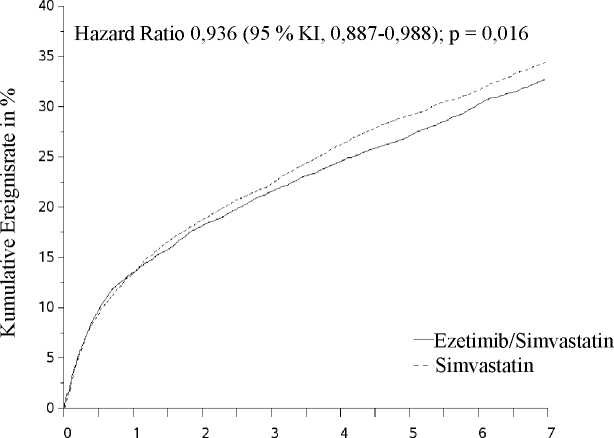
Randomisierung erfolgte koronare Revaskularisierung) und nicht-tödlicher Schlaganfall. Die Studie zeigte, dass eine Behandlung mit GOLTOR hinsichtlich der Reduktion von Ereignissen des primären kombinierten Endpunkts aus kardiovaskulärem Tod, schweren (major) koronaren Ereignissen (MCE) sowie nicht-tödlichem Schlaganfall im Vergleich zu einer Behandlung mit Simvastatin allein einen zusätzlichen Nutzen aufweist (relative Risikoreduktion um 6,4 %, p = 0,016). Der primäre Endpunkt trat bei 2.572 von 9.067 Patienten (Kaplan-Meier [KM] Ereignisrate nach 7 Jahren von 32,72 %) in der GOLTOR-Gruppe und bei 2.742 von 9.077 Patienten (Kaplan-Meier [KM] Ereignisrate nach 7 Jahren von 34,67 %) in der Simvastatin-Monotherapie-Gruppe auf (siehe Abbildung 1 und Tabelle 4). Die Gesamtsterblichkeit war in dieser Hochrisikogruppe unverändert (siehe Tabelle 1).
Insgesamt ergab sich ein Nutzen bei Betrachtung sämtlicher Schlaganfälle (unabhängig der Ursache), jedoch wurde ein geringer, nicht-signifikanter Anstieg hämorrhagischer Schlaganfälle in der Ezetimib/Simvastatin-Gruppe im Vergleich zur Simvastatin-Monotherapie-Gruppe beobachtet (siehe Tabelle 1). Das Risiko für hämorrhagischen Schlaganfall bei gemeinsamer Anwendung von Ezetimib mit einem stärker wirksamen Statin wurde im Rahmen von langfristigen Endpunktstudien nicht untersucht.
Die Wirkung der Behandlung mit Ezetimib/Simvastatin entsprach in vielen Subgruppen im Allgemeinen den Gesamtergebnissen, einschließlich Geschlecht, Alter, ethnische Herkunft, Diabetes mellitus in der Vorgeschichte, Ausgangslipidwerte, vorhergehende Statintherapie, vorangegangener Schlaganfall und Bluthochdruck.
Abbildung 1
Effekt von GOLTOR auf den primären kombinierten Endpunkt aus kardiovaskulärem Tod, schweren (major) koronaren Ereignissen (MCE) sowie nicht-tödlichem Schlaganfall
Time Since Randomization (Years)
Risikopatienten Ezetimib/Simvastatin 9067 Simvastatin 9077
7371
7455
6801
6799
6375
6327
5839
5729
4284 3301 1906
4206 3284 1857
Tabelle 4:
Schwere (major) kardiovaskuläre Ereignisse nach Behandlungsgruppe bei allen randomisierten Patienten der IMPROVE-IT-Studie
|
Outcome |
Ezetimib/Simvastatin 10 mg/40 mga (n = 9.067) |
Simvastatin 40 mgb (n = 9.077) |
Hazard Ratio (95 % KI) |
p-Wert | ||
|
n |
K-M %c |
n |
K-M %c | |||
|
Primärer kombinierter Wirksamkeitsendpunkt | ||||||
|
(Kardiovaskulärer Tod, schwere (major) koronare Ereignisse (MCE) und nicht-tödlicher Schlaganfall) |
2.572 |
32,72 % |
2.742 |
34,67 % |
0,936 (0,887; 0,988) |
0,016 |
|
Sekundäre kombinierte Wirksamkeitsendpunkte | ||||||
|
Tod durch KHK, nicht-tödlicher Myokardinfarkt, dringliche koronare Revaskularisierung nach 30 Tagen |
1.322 |
17,52 % |
1.448 |
18,88 % |
0,912 (0,847; 0,983) |
0,016 |
|
Schwere (major) koronare Ereignisse (MCE), nichttödlicher Schlaganfall, Tod (jegliche Ursache) |
3.089 |
38,65 % |
3.246 |
40,25 % |
0,948 (0,903; 0,996) |
0,035 |
|
Kardiovaskulärer Tod, nicht-tödlicher Myokardinfarkt, instabile Angina pectoris mit erforderlicher stationärer Einweisung, jegliche Revaskularisierung, nicht-tödlicher Schlaganfall |
2.716 |
34,49 % |
2.869 |
36,20 % |
0,945 (0,897; 0,996) |
0,035 |
Komponenten des primären kombinierten Endpunkts sowie ausgewählte Wirksamkeitsendpunkte
(erstmaliges Auftreten eines jeweiligen Ereignisses zu jeglichem Zeitpunkt)
|
Kardiovaskulärer Tod |
537 |
6,89 % |
538 |
6,84 % |
1,000 (0,887; 1,127) |
0,997 |
|
Schwere (major) koronare Ereignisse (MCE) | ||||||
|
Nicht-tödlicher Myokardinfarkt |
945 |
12,77 % |
1.083 |
14,41 % |
0,871 (0,798; 0,950) |
0,002 |
|
Instabile Angina pectoris mit erforderlicher stationärer Einweisung |
156 |
2,06 % |
148 |
1,92 % |
1,059 (0,846; 1,326) |
0,618 |
|
Koronare Revaskularisierung nach 30 Tagen |
1.690 |
21,84 % |
1.793 |
23,36 % |
0,947 (0,886; 1,012) |
0,107 |
|
Nicht-tödlicher Schlaganfall |
245 |
3,49 % |
305 |
4,24 % |
0,802 (0,678; 0,949) |
0,010 |
|
Myokardinfarkt (tödlich und nicht-tödlich) |
977 |
13,13 % |
1.118 |
14,82 % |
0,872 (0,800; 0,950) |
0,002 |
|
Schlaganfall (tödlich und nicht-tödlich) |
296 |
4,16 % |
345 |
4,77 % |
0,857 (0,734; 1,001) |
0,052 |
|
Nicht-hämorrhagischer Schlaganfall |
242 |
3,48 % |
305 |
4,23 % |
0,793 (0,670; 0,939) |
0,007 |
|
Hämorrhagischer Schlaganfall |
59 |
0,77 % |
43 |
0,59 % |
1,377 (0,930; 2,040) |
0,110 |
|
Tod jeglicher Ursache |
1.215 |
15,36 % |
1.231 |
15,28 % |
0,989 (0,914; 1,070) |
0,782 |
a 6 % wurden auf Ezetimib/Simvastatin 10 mg/80 mg hochtitriert b 27 % wurden auf Simvastatin 80 mg hochtitriert c Kaplan-Meier Schätzung nach 7 Jahren
d beinhaltet ischämischen Schlaganfall und nicht näher spezifizierten Schlaganfall
Primäre Hypercholesterinämie
In einer doppelblinden, plazebokontrollierten achtwöchigen Studie wurden 240 Patienten mit Hypercholesterinämie untersucht. Diese Patienten wurden bereits mit Simvastatin in Monotherapie behandelt, ohne das Ziel des National Cholesterol Education Program (NCEP) hinsichtlich des LDL-Cholesterinwerts zu erreichen (2,6-4,1 mmol/l [100-160 mg/dl], je nach Ausgangssituation). Sie wurden randomisiert und erhielten entweder 10 mg Ezetimib oder Plazebo zusätzlich zu ihrer laufenden Statin-Therapie. Von den Patienten, deren Ausgangswert für LDL-Cholesterin bei Studienbeginn den Zielwert unter Simvastatin-Therapie nicht erreicht hatte (etwa 80 %), erreichten bei Studienende unter Ezetimib mit Simvastatin signifikant mehr Patienten den LDL-Cholesterin-Zielwert (76 %) im Vergleich zu den Patienten unter Plazebo mit Simvastatin (21,5 %).
Die Unterschiede in den entsprechenden Senkungen des LDL-Cholesterins waren signifikant (27 % vs. 3 %). Außerdem senkte Ezetimib zusätzlich zu einer laufenden Statin-Therapie im Vergleich zu Plazebo signifikant die Werte von Gesamtcholesterin, Apolipoprotein B und Triglyzeriden.
In eine multizentrische, doppelblinde, 24-wöchige Studie wurden 214 Patienten mit Typ-2-Diabetes mellitus eingeschlossen; sie wurden mit Thiazolidindionen (Rosiglitazon oder Pioglitazon) mindestens 3 Monate und mit 20 mg Simvastatin mindestens 6 Wochen behandelt und ihr LDL-Cholesterinwert
lag im Mittel bei 2,4 mmol/l (93 mg/dl). Sie wurden randomisiert und erhielten entweder 40 mg Simvastatin oder die Wirkstoffe, die GOLTOR 10 mg/20 mg entsprechen, in Koadministration. GOLTOR 10 mg/20 mg war signifikant wirksamer als die Verdopplung der Simvastatindosis auf 40 mg bei der weiteren Senkung der Werte von LDL-Cholesterin (-21 % vs. 0 %), Gesamtcholesterin (-14 % vs. -1 %), Apolipoprotein B (-14 % vs. -2 %) und non-HDL-Cholesterin (-20 % vs. -2 %) über die bereits mit 20 mg Simvastatin erreichten Senkungen hinaus. Die Ergebnisse hinsichtlich der Werte von HDL-Cholesterin und Triglyzeriden unterschieden sich nicht signifikant zwischen beiden Behandlungsgruppen. Die Ergebnisse wurden nicht durch die Wahl des Thiazolidindions beeinflusst.
Die Wirksamkeit der verschiedenen Stärken von GOLTOR (10 mg/10 mg bis 10 mg/80 mg pro Tag) wurde in einer multizentrischen, doppelblinden, plazebokontrollierten, zwölfwöchigen Studie untersucht, die alle verfügbaren Dosierungen von GOLTOR und alle relevanten Dosierungen von Simvastatin umfasste. Ein Vergleich der Patienten unter allen Dosen von GOLTOR mit denen unter allen Dosen von Simvastatin zeigte, dass GOLTOR signifikant stärker die Werte von Gesamtcholesterin, LDL-Cholesterin und Triglyzeriden (siehe Tabelle 5) sowie von Apolipoprotein B (-42 % vs. -29 %), non-HDL-Cholesterin (-49 % vs. -34 %) und C-reaktivem Protein (-33 % vs. -9 %) senkte. Die Wirkungen von GOLTOR auf die Werte von HDL-Cholesterin waren den unter Simvastatin beobachteten Werten ähnlich. Eine weitere Analyse zeigte, dass GOLTOR die Werte von HDL-Cholesterin im Vergleich zu Plazebo signifikant erhöhte.
Tabelle 5:
Das Ansprechen auf GOLTOR von Patienten mit primärer Hypercholesterinämie (mittlerea) Veränderung in % zum Ausgangswert [unbehandelt]b))
|
Therapie (Tagesdosis) |
N |
Gesamt- cholesteri |
LDL- Cholesteri |
HDL- Cholesteri |
Triglyzeride a) |
|
n |
n |
n | |||
|
Gepoolte Daten (alle Dosen von GOLTOR)c) |
353 |
-38 |
-53 |
+8 |
-28 |
|
Gepoolte Daten (alle Dosen von Simvastatin)c) |
349 |
-26 |
-38 |
+8 |
-15 |
|
Ezetimib 10 mg |
92 |
-14 |
-20 |
+7 |
-13 |
|
Plazebo |
93 |
+2 |
+3 |
+2 |
-2 |
|
GOLTOR pro Dosis 10 mg/10 mg |
87 |
-32 |
-46 |
+9 |
-21 |
|
10 mg/20 mg |
86 |
-37 |
-51 |
+8 |
-31 |
|
10 mg/40 mg |
89 |
-39 |
-55 |
+9 |
-32 |
|
10 mg/80 mg |
91 |
-43 |
-61 |
+6 |
-28 |
|
Simvastatin pro Dosis 10 mg |
81 |
-21 |
-31 |
+5 |
-4 |
|
20 mg |
90 |
-24 |
-35 |
+6 |
-14 |
|
40 mg |
91 |
-29 |
-42 |
+8 |
-19 |
|
80 mg |
87 |
-32 |
-46 |
+11 |
-26 |
a) Für Triglyzeride % Median-Veränderung vom Ausgangswert
b) Ausgangswert - keine Behandlung mit einem lipidsenkenden Arzneimittel
c) Gepoolte Dosen von GOLTOR (10 mg/10 mg - 10 mg/80 mg) senkten im Vergleich zu Simvastatin signifikant Werte von Gesamtcholesterin, LDL-Cholesterin und Triglyzeriden und erhöhten die Werte von HDL-Cholesterin signifikant im Vergleich zu Plazebo.
In einer Studie mit ähnlichem Design waren die Ergebnisse für alle Lipidparameter im Allgemeinen übereinstimmend. In einer gepoolten Analyse beider Studien war bei Patienten mit Triglyzeridwerten über oder unter 200 mg/dl das Ansprechen hinsichtlich der Lipidwerte auf GOLTOR vergleichbar.
In einer multizentrischen, doppelblinden, kontrollierten klinischen Studie (ENHANCE) erhielten 720 Patienten mit heterozygoter familiärer Hypercholesterinämie 2 Jahre lang randomisiert entweder 10 mg Ezetimib in Kombination mit 80 mg Simvastatin (n = 357) oder 80 mg Simvastatin (n = 363).
Das primäre Ziel der Studie war, die Wirkung der Ezetimib/Simvastatin-Kombinationstherapie im Vergleich zur Simvastatin-Monotherapie auf die Dicke der Intima media (intima-media thickness [IMT]) der A. carotis zu untersuchen. Die Bedeutung dieses Surrogatmarkers für die kardiovaskuläre Morbidität und Mortalität ist noch unklar.
Der primäre Endpunkt, die Änderung der mittleren IMT aller sechs Karotissegmente unterschied sich in den Ultraschall-B-Messungen nicht signifikant (p = 0,29) zwischen beiden Behandlungsgruppen.
10 mg Ezetimib in Kombination mit 80 mg Simvastatin begrenzte während der 2-jährigen Studiendauer die IMT-Verdickung auf 0,0111 mm, Simvastatin 80 mg allein auf 0,0058 mm (mittlerer Ausgangswert der IMT der A. carotis entsprach 0,68 mm bzw. 0,69 mm).
10 mg Ezetimib in Kombination mit 80 mg Simvastatin senkte die Werte von LDL-Cholesterin, Gesamtcholesterin, Apo-B und Triglyzeriden signifikant stärker als 80 mg Simvastatin allein. Die prozentuale Erhöhung des HDL-Cholesterins war in beiden Behandlungsgruppen ähnlich. Die Nebenwirkungen, die unter der Kombination von 10 mg Ezetimib mit 80 mg Simvastatin berichtet wurden, entsprachen dem bekannten Sicherheitsprofil.
GOLTOR enthält Simvastatin. In zwei groß angelegten, plazebokontrollierten klinischen Studien, der Scandinavian Simvastatin Survival Study (20 mg-40 mg; n = 4.444 Patienten) und der Heart Protection Study (40 mg; n = 20.536 Patienten), wurden die Wirkungen einer Simvastatin-Therapie bei für koronare Ereignisse hochgefährdeten Patienten untersucht. Die Patienten litten entweder an einer KHK, Diabetes, peripheren Gefäßerkrankungen oder hatten bereits einen Schlaganfall oder ein anderes koronares Ereignis in der Krankengeschichte. Simvastatin führt nachweislich zu einer Reduktion des Gesamtmortalitätsrisikos aufgrund der Reduktion KHK-bedingter Todesfälle, des Risikos nicht letaler Myokardinfarkte und Schlaganfälle sowie der Notwendigkeit koronarer und nicht koronarer revaskularisierender Eingriffe.
Die SEARCH-Studie (Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine) verglich die Wirkung der Behandlung von 80 mg Simvastatin mit der von 20 mg Simvastatin (mediane Nachbeobachtung von 6,7 Jahren) auf schwere vaskuläre Ereignisse (MVE, major vascular events, definiert als KHK mit Todesfolge, nicht-tödlicher Myokardinfarkt, koronare Revaskularisierungseingriffe, nicht-tödlicher Schlaganfall oder Schlaganfall mit Todesfolge, periphere Revaskularisierungseingriffe) bei 12.064 Patienten mit einem Myokardinfarkt in der Krankengeschichte. Es zeigte sich kein signifikanter Unterschied zwischen beiden Gruppen hinsichtlich dieser Ereignisse: 20 mg Simvastatin (n = 1.553; 25,7 %) vs. 80 mg Simvastatin (n = 1.477; 24,5 %); RR 0,94, 95 % KI: 0,88 - 1,01. Der absolute Unterschied der LDL-Cholesterinwerte zwischen beiden Gruppen betrug im Verlauf der Studie 0,35 ± 0,01 mmol/l. Das Sicherheitsprofil war ebenfalls bei beiden Behandlungsgruppen ähnlich, mit der Ausnahme der Myopathiehäufigkeit, die bei Patienten unter 80 mg Simvastatin ca. 1,0 % und im Vergleich dazu bei Patienten unter 20 mg Simvastatin 0,02 % betrug. Etwa die Hälfte dieser Myopathiefälle ereignete sich im ersten Jahr der Behandlung. Die Myopathiehäufigkeit in den folgenden Jahren lag bei jeweils ca.
0,1 %.
Kinder und Jugendliche
In einer multizentrischen, doppelblinden, kontrollierten Studie wurden 142 Jungen (Tanner-Stadium II und darüber) und 106 Mädchen nach der Menarche im Alter von 10 bis 17 Jahren (mittleres Alter
14,2 Jahre) mit heterozygoter familiärer Hypercholesterinämie (HeFH) und LDL-Cholesterin-Ausgangswerten von 4,1 bis 10,4 mmol/l (159 bis 402 mg/dl) untersucht. Sie erhielten randomisiert 6 Wochen entweder Ezetimib 10 mg mit Simvastatin (10, 20 oder 40 mg) oder Simvastatin allein (10, 20 oder 40 mg), danach 27 Wochen Ezetimib zusammen mit 40 mg Simvastatin oder 40 mg Simvastatin allein sowie im Anschluss in einer offenen Studienverlängerung 20 Wochen Ezetimib mit Simvastatin (10 mg, 20 mg oder 40 mg).
Nach 6 Wochen führte die gemeinsame Gabe von Ezetimib und Simvastatin (alle Dosen) zu signifikant niedrigeren Werten von Gesamtcholesterin (38 % vs. 26 %), LDL-Cholesterin (49 % vs.
34 %), Apo-B (39 % vs. 27 %) und non-HDL-Cholesterin (47 % vs. 33 %) als Simvastatin (alle Dosen) allein. Die Ergebnisse der Triglyzeridwerte (-17 % vs. -12 %) und HDL-Cholesterin (+7 % vs.
+6 %) waren in beiden Behandlungsgruppen ähnlich. Nach 33 Wochen stimmten die Ergebnisse mit den Werten nach 6 Wochen überein, wobei signifikant mehr Patienten unter Ezetimib zusammen mit 40 mg Simvastatin (62 %) das gemeinsame Behandlungsziel des NCEP („National Cholesterol Education Program“) und der AAP („American Academy of Pediatrics“) (< 2,8 mmol/l [110 mg/dl]) für LDL-Cholesterin als jene Patienten unter 40 mg Simvastatin (25 %) erreichten. Nach 53 Wochen, am Ende der offenen Studienverlängerung, blieben die Wirkungen auf die Lipidwerte konstant.
Sicherheit und Wirksamkeit von Ezetimib mit Simvastatin in Dosen über 40 mg pro Tag wurden bei pädiatrischen Patienten im Alter von 10 bis 17 Jahren nicht untersucht. Die Langzeitwirkung der Therapie mit Ezetimib bei Patienten unter 17 Jahren auf die Reduktion von Morbidität und Mortalität im Erwachsenenalter wurde nicht untersucht.
Die Europäische Arzneimittel-Agentur hat für GOLTOR eine Freistellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in allen pädiatrischen Altersklassen im Anwendungsgebiet Hypercholesterinämie gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen).
Homozygote familiäre Hypercholesterinämie
In einer doppelblinden, randomisierten, zwölfwöchigen Studie wurden Patienten mit klinischer und/oder genotypischer Diagnose einer homozygoten familiären Hypercholesterinämie untersucht. Daten einer Subgruppe mit Patienten (n = 14), die zu Studienbeginn 40 mg Simvastatin erhielten, wurden analysiert. Eine Dosiserhöhung von 40 mg auf 80 mg Simvastatin (n = 5) führte zu einer Senkung des LDL-Cholesterinwerts um 13 % im Vergleich zum Ausgangswert unter 40 mg Simvastatin. Eine Koadministration von Ezetimib und Simvastatin entsprechend GOLTOR (10 mg/40 mg und 10 mg/80 mg gepoolt, n = 9) führte zu einer Senkung des LDL-Cholesterinwerts um 23 % im Vergleich zum Ausgangswert unter 40 mg Simvastatin. Bei jenen Patienten führte die Koadministration von Ezetimib und Simvastatin entsprechend GOLTOR (10 mg/80 mg, n = 5) zu einer Senkung des LDL-Cholesterinwerts um 29 % im Vergleich zum Ausgangswert unter 40 mg Simvastatin.
Prävention schwerer (major) vaskulärer Ereignisse bei chronischer Nierenerkrankung Die SHARP-Studie (Study of Heart and Renal Protection) war eine multinationale, randomisierte, plazebokontrollierte, doppelblinde Studie mit 9.438 Patienten mit chronischer Nierenerkrankung, wobei ein Drittel der Patienten bereits zu Studienbeginn dialysepflichtig war. Insgesamt wurden 4.650 Patienten der GOLTOR 10 mg/20 mg Gruppe und 4.620 Patienten der Plazebo-Gruppe zugewiesen und über einen medianen Zeitraum von 4,9 Jahren beobachtet. Die Patienten waren im Mittel 62 Jahre alt, 63 % waren Männer, 72 % waren kaukasischer Abstammung, 23 % waren Diabetiker. Die nicht-dialysepflichtigen Patienten hatten eine mittlere geschätzte glomeruläre Filtrationsrate (eGFR) von 26,5 ml/min/1,73 m2. Es gab keine Einschlusskriterien hinsichtlich der Lipidparameter. Zu Studienbeginn betrug die mittlere LDL-Cholesterin-Konzentration 108 mg/dl. Einschließlich der Patienten, die keine Studienmedikation mehr einnahmen, betrug nach einem Jahr Behandlungsdauer die Reduktion des LDL-Cholesterinspiegels relativ zu Plazebo 26 % bei alleiniger Gabe von 20 mg Simvastatin und 38 % unter GOLTOR 10 mg/20 mg.
Der im Studienprotokoll der SHARP-Studie festgelegte primäre Vergleich war eine Intention-to-treat (ITT)-Analyse „schwerer (major) vaskulärer Ereignisse“ („MVE“; definiert als nicht-tödlicher Myokardinfarkt [MI] oder Herztod, Schlaganfall oder jegliche Behandlung zur Revaskularisierung) bei ausschließlich den Patienten, die anfänglich in die GOLTOR (n = 4.193) oder Plazebo (n = 4.191) Studiengruppe randomisiert wurden. Die Sekundäranalysen schlossen sowohl die Untersuchung des kombinierten Endpunktes (MVE) als auch die darin enthaltenen einzelnen Endpunkte über die gesamte auf GOLTOR (n = 4.650) oder Plazebo (n = 4.620) randomisierte Studienpopulation (zu Studienbeginn oder zum Zeitpunkt nach 1 Jahr) ein.
Die primäre Endpunktanalyse zeigte, dass GOLTOR das Risiko von schweren (major) vaskulären Ereignissen signifikant mit einer relativen Risikoreduktion von 16 % (p = 0,001) senkte (749 Patienten mit Ereignissen in der Plazebo-Gruppe versus 639 Patienten in der GOLTOR Gruppe).
Allerdings ermöglichte das Studiendesign keine Aussage zum Beitrag des Einzelbestandteils Ezetimib zur Wirksamkeit im Sinne einer signifikanten Risikoreduktion von schweren (major) vaskulären Ereignissen bei Patienten mit chronischer Nierenerkrankung.
Die einzelnen Komponenten des MVE-Kombinationsereignisses bei allen randomisierten Patienten sind in Tabelle 6 dargestellt. GOLTOR reduzierte signifikant das Risiko eines Schlaganfalls und jeglicher Revaskularisierung. Bezüglich nicht-tödlichem Myokardinfarkt und Herztod besteht ein nicht signifikanter, numerischer Vorteil von GOLTOR gegenüber Plazebo.
Tabelle 6:
Schwere (major) vaskuläre Ereignisse bei allen in SHARPa) randomisierten Patienten
aufgeführt nach Behandlungsgruppe
|
Outcome |
GOLTOR 10 mg/20 mg (n = 4.650) |
Plazebo (n = 4.620) |
Relatives Risiko (95 % KI) |
p-Wert |
|
Schwere (major) vaskuläre |
701 (15,1 %) |
814 (17,6 %) |
0,85 (0,77-0,94) |
0,001 |
|
Ereignisse (MVE) | ||||
|
Nicht-tödlicher MI |
134 (2,9 %) |
159 (3,4 %) |
0,84 (0,66-1,05) |
0,12 |
|
Herztod |
253 (5,4 %) |
272 (5,9 %) |
0,93 (0,78-1,10) |
0,38 |
|
Jeglicher Schlaganfall |
171 (3,7 %) |
210 (4,5 %) |
0,81 (0,66-0,99) |
0,038 |
|
Nicht-hämorrhagischer |
131 (2,8 %) |
174 (3,8 %) |
0,75 (0,60-0,94) |
0,011 |
|
Schlaganfall | ||||
|
Hämorrhagischer |
45 (1,0 %) |
37 (0,8 %) |
1,21 (0,78-1,86) |
0,40 |
|
Schlaganfall | ||||
|
Jegliche Revaskularisierung |
284 (6,1 %) |
352 (7,6 %) |
0,79 (0,68-0,93) |
0,004 |
|
Schwere (major) |
526 (11,3 %) |
619 (13,4 %) |
0,83 (0,74-0,94) |
0,002 |
|
atherosklerotische Ereignisse (MAE)b) | ||||
a) Intention-to-treat-Analyse aller Patienten, welche in der SHARP-Studie in die Behandlungsarme GOLTOR oder Plazebo randomisiert wurden (initial oder nach einem Jahr)
b) MAE: definiertes Kombinationsereignis einschließlich nicht-tödlicher Myokardinfarkt (MI), koronar bedingter Tod, nicht-hämorrhagischer Schlaganfall oder jegliche Revaskularisierung
Die mit GOLTOR erzielte absolute Senkung von LDL-Cholesterin war bei den Patienten, die zu Anfang der Studie niedrige LDL-Cholesterinspiegel (< 2,5 mmol/l) hatten oder bei Dialysepatienten im Vergleich zu den übrigen Patienten geringer. Die Risikoreduktion dieser beiden Patientengruppen war entsprechend vermindert.
Aortenstenose
Die SEAS (Simvastatin and Ezetimibe for the Treatment of Aortic Stenosis) Studie war eine multizentrische, doppelblinde, plazebokontrollierte Studie mit einer medianen Dauer von 4,4 Jahren, an der 1.873 Patienten mit asymptomatischer Aortenstenose (AS) - dokumentiert durch mit Dopplermessungen ermittelte maximale Fließgeschwindigkeit („Peakflow“) in der Aorta im Bereich von 2,5 bis 4,0 m/s - teilnahmen. Es wurden nur Patienten eingeschlossen, für die keine Statinbehandlung zur Reduktion des Risikos einer atherosklerotischen kardiovaskulären Erkrankung als notwendig erachtet wurde. Die Patienten wurden 1:1 randomisiert und erhielten entweder Plazebo oder eine Kombination aus 10 mg Ezetimib und 40 mg Simvastatin täglich.
Der primäre Endpunkt war eine Kombination schwerer kardiovaskulärer Ereignisse (major cardiovascular events [MCE]) und umfasste kardiovaskulären Tod, chirurgischen Ersatz einer Aortenklappe (aortic valve replacement [AVR]), Herzinsuffizienz (congestive heart failure [CHF]) als Folge einer fortschreitenden AS, nicht-tödlichen Myokardinfarkt, Einsetzen eines aorto-koronaren Bypasses (coronary artery bypass grafting [CABG]), perkutane koronare Intervention (percutaneous coronary intervention [PCI]), Krankenhauseinweisung wegen instabiler Angina pectoris und nichthämorrhagischem Schlaganfall. Die wichtigsten sekundären Endpunkte waren aus Untergruppen der Kategorien der Ereignisse des primären Endpunkts zusammengesetzt.
Im Vergleich zu Plazebo führte die Kombination von 10 mg Ezetimib und 40 mg Simvastatin nicht zu einer signifikanten Verringerung des Risikos für MCE.
Der primäre Endpunkt trat bei 333 Patienten (35,3 %) in der Ezetimib/Simvastatin-Gruppe und bei 355 Patienten (38,2 %) in der Plazebo-Gruppe auf („hazard ratio“ in der Ezetimib/Simvastatin-Gruppe, 0,96; 95 % Konfidenzintervall, 0,83 bis 1,12; p = 0,59). Die Aortenklappe wurde bei 267 Patienten (28,3 %) in der Ezetimib/Simvastatin-Gruppe und bei 278 Patienten (29,9 %) in der Plazebo-Gruppe („hazard ratio“, 1,00; 95 % KI, 0,84 bis 1,18; p = 0,97) ersetzt. Ein ischämisches kardiovaskuläres Ereignis erlitten weniger Patienten in der Ezetimib/Simvastatin-Gruppe (n = 148) als in der Plazebo-Gruppe (n = 187) („hazard ratio“, 0,78; 95 % KI, 0,63 bis 0,97; p = 0,02), hauptsächlich aufgrund der kleineren Anzahl an Patienten, die einen Koronarbypass erhielten.
Es kam in der Ezetimib/Simvastatin-Gruppe häufiger zu Krebsfällen (105 versus 70, p = 0,01). Die klinische Bedeutung dieser Beobachtung ist unklar, da sich in der größeren SHARP-Studie die Gesamtzahl der Krebsfälle nicht unterschied (438 in der Ezetimib/Simvastatin-Gruppe versus 439 in der Plazebo-Gruppe). Darüber hinaus unterschied sich in der IMPROVE-IT-Studie die Gesamtzahl der neu diagnostizierten Krebserkrankungen nicht signifikant zwischen den Behandlungsgruppen (853 in der Ezetimib/Simvastatin-Gruppe im Vergleich zu 863 in der Simvastatin-Monotherapie-Gruppe), so dass die Beobachtung aus der SEAS-Studie weder durch die SHARP-Studie noch durch die IMPROVE-IT-Studie bestätigt werden konnte.
5.2 Pharmakokinetische Eigenschaften
Bei der gemeinsamen Gabe von Ezetimib und Simvastatin wurde keine signifikante pharmakokinetische Interaktion beobachtet.
Resorption
GOLTOR
GOLTOR ist bioäquivalent zur Koadministration von Ezetimib und Simvastatin.
Ezetimib
Nach oraler Gabe wird Ezetimib rasch resorbiert und weitgehend zu einem pharmakologisch aktiven Phenol-Glukuronid (Ezetimib-Glukuronid) konjugiert. Die mittlere Plasmaspitzenkonzentration (Cmax) wird nach 1-2 Stunden für Ezetimib-Glukuronid und nach 4-12 Stunden für Ezetimib erreicht. Die absolute Bioverfügbarkeit von Ezetimib kann nicht bestimmt werden, da die Substanz in wässrigen Lösungen, welche zur Injektion geeignet sind, praktisch unlöslich ist.
Eine gleichzeitige Nahrungsaufnahme (Mahlzeiten mit hohem Fettgehalt oder fettfreie Mahlzeiten) hatte keinen Einfluss auf die orale Bioverfügbarkeit von Ezetimib, wenn es in der Form von 10-mg-Tabletten angewendet wurde.
Simvastatin
Die Verfügbarkeit der aktiven Betahydroxysäure nach einer oralen Simvastatindosis betrug im systemischen Kreislauf weniger als 5 % der Dosis, was mit dem ausgeprägten First-Pass-Effekt in der Leber übereinstimmt. Im menschlichen Plasma sind der Betahydroxysäure-Metabolit und 4 weitere aktive Metaboliten als Hauptmetaboliten von Simvastatin zu finden.
Das Plasmaprofil der aktiven als auch gesamten Inhibitoren wurde im Vergleich zum Nüchternzustand nicht beeinflusst, wenn Simvastatin unmittelbar vor einer Testmahlzeit eingenommen wurde.
Verteilung
Ezetimib
Ezetimib ist beim Menschen zu 99,7 %, Ezetimib-Glukuronid zu 88-92 % an Plasmaproteine gebunden.
Simvastatin
Simvastatin und die Betahydroxysäure sind beim Menschen an Plasmaproteine gebunden (95 %).
Die Ergebnisse pharmakokinetischer Studien bei einmaliger und multipler Gabe von Simvastatin zeigten, dass die wiederholte Verabreichung des Arzneimittels nicht zu einer Akkumulation führt. In allen oben erwähnten pharmakokinetischen Studien traten die maximalen Inhibitorkonzentrationen im Plasma zwischen 1,3 und 2,4 Stunden nach der Einnahme auf.
Biotransformation
Ezetimib
Ezetimib wird vor allem im Dünndarm und in der Leber über Glukuronidkonjugation (eine Phase-II-Reaktion) metabolisiert und anschließend über die Galle ausgeschieden. In allen untersuchten Spezies wurde ein minimaler oxidativer Metabolismus (eine Phase-I-Reaktion) beobachtet. Ezetimib und Ezetimib-Glukuronid sind die hauptsächlich im Plasma nachgewiesenen Substanzen, wobei Ezetimib ca. 10-20 % und Ezetimib-Glukuronid ca. 80-90 % der Gesamtkonzentration im Plasma ausmachen. Ezetimib und Ezetimib-Glukuronid werden langsam aus dem Plasma eliminiert mit Hinweis auf einen signifikanten enterohepatischen Kreislauf. Die Halbwertszeit von Ezetimib und Ezetimib-Glukuronid beträgt ca. 22 Stunden.
Simvastatin
Simvastatin, ein inaktives Lacton, wird in vivo schnell zur entsprechenden Betahydroxysäure, einem potenten Hemmer der HMG-CoA-Reduktase, hydrolysiert. Die Hydrolyse findet vor allem in der Leber statt; im menschlichen Plasma verläuft sie sehr langsam.
Beim Menschen wird Simvastatin gut resorbiert und unterliegt einem ausgeprägten First-Pass-Effekt in der Leber - abhängig vom Blutfluss in der Leber. Die Leber ist der primäre Wirkort des Medikaments mit nachfolgender Ausscheidung von Arzneimitteläquivalenten über die Galle. Daraus resultiert eine niedrige Verfügbarkeit der wirksamen Substanz im systemischen Kreislauf.
Die Halbwertszeit des Betahydroxysäure-Metaboliten beträgt durchschnittlich 1,9 Stunden nach intravenöser Injektion.
Elimination
Ezetimib
Nach oraler Gabe einer radioaktiv markierten Dosis von 20 mg [14C]Ezetimib an Probanden finden sich ca. 93 % der gesamten Radioaktivität im Plasma als Gesamt-Ezetimib. Über einen Beobachtungszeitraum von 10 Tagen wurden ca. 78 % der gegebenen radioaktiven Dosis in den Fäzes und 11 % im Urin wiedergefunden. Nach 48 Stunden war keine Radioaktivität mehr im Plasma nachweisbar.
Simvastatin
Simvastatinsäure wird durch den Transporter OATP1B1 aktiv in die Hepatozyten aufgenommen.
Nach oraler Gabe radioaktiv markierten Simvastatins an Probanden wurden innerhalb von 96 Stunden 13 % der Radioaktivität im Urin und 60 % in den Fäzes wiedergefunden. Letztere Menge steht sowohl für resorbierte Anteile, die über die Galle ausgeschieden werden, als auch für nicht resorbierte Substanz. Nur durchschnittlich 0,3 % einer i.v.-Dosis wurden nach intravenöser Injektion des Betahydroxysäure-Metaboliten im Urin als Inhibitoren ausgeschieden.
Besondere Patientengruppen
Kinder und Jugendliche
Resorption und Metabolismus von Ezetimib sind bei Kindern und Jugendlichen (10-18 Jahre) ähnlich wie bei Erwachsenen. Die Pharmakokinetik des Gesamt-Ezetimibs bei Jugendlichen und Erwachsenen unterscheidet sich nicht. Pharmakokinetische Daten für Kinder unter 10 Jahren liegen noch nicht vor. Klinische Erfahrungen zur Behandlung von Kindern und Jugendlichen umfassen Patienten mit homozygoter familiärer Hypercholesterinämie, heterozygoter familiärer Hypercholesterinämie oder Sitosterinämie (siehe Abschnitt 4.2).
Ältere Patienten
Die Plasmakonzentrationen von Gesamt-Ezetimib sind bei älteren Patienten (ab 65 Jahren) etwa doppelt so hoch wie bei jüngeren Patienten (18-45 Jahre). Die Senkung des LDL-Cholesterinwerts und das Sicherheitsprofil sind jedoch bei älteren und jüngeren mit Ezetimib behandelten Probanden vergleichbar (siehe Abschnitt 4.2).
Einschränkung der Leberfunktion
Nach einer Einzeldosis von 10 mg Ezetimib bei Patienten mit leichter Einschränkung der Leberfunktion (Child-Pugh-Score 5 oder 6) war die AUC für Gesamt-Ezetimib ca. 1,7-mal größer als jene für gesunde Probanden. In einer 14-tägigen Studie mit Mehrfachdosierungen (10 mg pro Tag) bei Patienten mit mäßiger Einschränkung der Leberfunktion (Child-Pugh-Score 7-9) war die mittlere AUC für Gesamt-Ezetimib am 1. und am 14. Tag ca. 4-mal größer als die von gesunden Probanden. Für Patienten mit leichter Einschränkung der Leberfunktion ist keine Dosisanpassung erforderlich. Da die Folgen einer erhöhten Exposition mit Gesamt-Ezetimib bei Patienten mit mäßiger oder mit schwerer Einschränkung der Leberfunktion (Child-Pugh-Score > 9) nicht bekannt sind, wird Ezetimib für diese Patienten nicht empfohlen (siehe Abschnitte 4.2 und 4.4).
Einschränkung der Nierenfunktion
Ezetimib
Nach einer Einzeldosis von 10 mg Ezetimib bei Patienten mit schwerer Nierenerkrankung (n = 8; mittlere Kreatinin-Clearance < 30 ml/min) war die mittlere AUC für Gesamt-Ezetimib im Vergleich zu der bei gesunden Probanden (n = 9) um das ca. 1,5-Fache vergrößert (siehe Abschnitt 4.2).
Ein Patient in dieser Studie (nach Nierentransplantation, unter multipler Arzneimitteltherapie, u. a. Ciclosporin) hatte eine 12fach höhere Exposition mit Gesamt-Ezetimib.
Simvastatin
In einer klinischen Studie zeigte sich bei Patienten mit schwerer Einschränkung der Nierenfunktion (Kreatinin-Clearance < 30 ml/min), dass nach Einmalgabe von einem eng verwandten HMG-CoA-Reduktase-Inhibitor die Plasmaspiegel der Gesamtinhibitoren etwa zweimal höher waren als bei gesunden Probanden.
Geschlecht
Die Plasmakonzentrationen von Gesamt-Ezetimib sind bei Frauen etwas höher (ca. 20 %) als bei Männern. Unter Therapie mit Ezetimib sind sowohl die Senkung des LDL-Cholesterinwerts als auch das Sicherheitsprofil bei Männern und Frauen vergleichbar.
SLCO1B1-Polymorphismus
Träger des c.521T>C-Allels des SLCO1B1-Gens haben eine niedrigere OATP1B1-Aktivität. Die durchschnittliche Bioverfügbarkeit (AUC) des wichtigsten aktiven Metaboliten, Simvastatinsäure, beträgt 120 % bei heterozygoten Trägern (CT) des C-Allels und 221 % bei homozygoten Trägern (CC), bezogen auf die von Patienten mit dem häufigsten Genotyp (TT). Das C-Allel hat eine Häufigkeit von 18 % in der europäischen Bevölkerung. Bei Patienten mit SLCO1B1-Polymorphismus besteht ein Risiko für eine verstärkte Exposition von Simvastatinsäure, welche zu einem erhöhten Rhabdomyolyserisiko führen kann (siehe Abschnitt 4.4).
5.3 Präklinische Daten zur Sicherheit
GOLTOR
In Koadministrationsstudien mit Ezetimib und Simvastatin wurden im Wesentlichen die toxischen Effekte beobachtet, die für die Behandlung mit Statinen typisch sind. Manche toxischen Effekte waren stärker ausgeprägt als bei Monotherapie mit Statinen. Dieses wird auf pharmakokinetische und/oder pharmakodynamische Interaktionen bei Koadministrationsbehandlung zurückgeführt. Derartige Interaktionen traten in den klinischen Studien nicht auf. Myopathien traten bei Ratten nur bei Exposition mit Dosen auf, die um ein Vielfaches über der humantherapeutischen Dosis lagen (ca. 20fache AUC-Konzentration für Simvastatin und 1.800fache AUC-Konzentration für die aktiven Metaboliten). Es fanden sich keine Hinweise auf einen Einfluss der gemeinsamen Gabe von Ezetimib auf das myotoxische Potenzial von Simvastatin.
Bei Hunden wurden unter Koadministration von Ezetimib und Simvastatin einige Auswirkungen auf die Leber bei niedrigen Expositionen (< fache AUC beim Menschen) beobachtet. Die Leberenzyme (ALT, AST) stiegen ohne Anzeichen einer Gewebsnekrose deutlich an. Histopathologische Befunde (Gallengangshyperplasie, Pigmentakkumulation, mononukleäre Zellinfiltrate und verkleinerte Hepatozyten, „small hepatocytes“) wurden bei Hunden unter Koadministration von Ezetimib und Simvastatin beobachtet. Diese Veränderungen waren bei einer Behandlungsdauer bis zu 14 Monaten nicht progredient. Nach Absetzen der Behandlung waren die Leberbefunde allgemein reversibel. Diese Ergebnisse stimmten mit den für HMG-CoA-Reduktase-Inhibitoren beschriebenen Befunden überein oder wurden den sehr niedrigen Cholesterinwerten zugeschrieben, die bei den betroffenen Hunden erreicht wurden.
Die gleichzeitige Gabe von Ezetimib und Simvastatin war bei Ratten nicht teratogen. Bei trächtigen Kaninchen wurde eine geringe Anzahl von Skelettmissbildungen (Blockwirbelbildung und verminderte Anzahl an Schwanzwirbeln) beobachtet.
In einer Reihe von In-vivo- und In-vitro-Assays zeigte Ezetimib allein oder zusammen mit Simvastatin kein genotoxisches Potenzial.
Ezetimib
In Tierstudien zur chronischen Toxizität von Ezetimib wurden keine Zielorgane für toxische Wirkungen identifiziert. Bei Hunden war nach 4-wöchiger Behandlung mit Ezetimib (> 0,03 mg/kg/Tag) die Cholesterinkonzentration in der Blasengalle um das 2,5- bis 3,5-Fache erhöht. In einer Studie an Hunden über ein Jahr wurde bei Dosen bis zu 300 mg/kg/Tag jedoch keine erhöhte Inzidenz von Cholelithiasis oder anderen hepatobiliären Effekten beobachtet. Die Bedeutung dieser Daten für den Menschen ist nicht bekannt. Ein lithogenes Risiko bei der therapeutischen Anwendung von Ezetimib kann nicht ausgeschlossen werden.
Langzeitstudien zur Kanzerogenität von Ezetimib verliefen negativ.
Ezetimib hatte weder einen Einfluss auf die Fertilität von männlichen oder weiblichen Ratten, noch erwies es sich bei Ratten und Kaninchen als teratogen, auch beeinflusste es nicht die prä- oder postnatale Entwicklung. Ezetimib war bei trächtigen Ratten und Kaninchen unter multiplen Dosen von
1.000 mg/kg/Tag plazentagängig.
Simvastatin
Basierend auf konventionellen Tierstudien zu Pharmakodynamik, Toxizität bei wiederholt verabreichten Dosen, Genotoxizität und Kanzerogenität lassen sich keine Risiken für den Patienten ableiten, die nicht aufgrund des pharmakologischen Mechanismus zu erwarten wären. In den höchsten von Ratte und Kaninchen vertragenen Dosen rief Simvastatin keine fetalen Missbildungen hervor und hatte keine Auswirkungen auf Fertilität, Fortpflanzung oder neonatale Entwicklung.
6. PHARMAZEUTISCHE ANGABEN
6.1 Liste der sonstigen Bestandteile
Butylhydroxyanisol (Ph. Eur.)
Citronensäure-Monohydrat Croscarmellose-Natrium Hypromellose Lactose -Monohydrat Magnesiumstearat (Ph. Eur.)
Mikrokristalline Cellulose Propylgallat (Ph. Eur.)
6.2 Inkompatibilitäten
Nicht zutreffend.
6.3 Dauer der Haltbarkeit
2 Jahre
6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Nicht über 30 °C lagern.
Blister: In der Originalverpackung aufbewahren, um das Arzneimittel vor Feuchtigkeit und Licht zu schützen.
6.5 Art und Inhalt des Behältnisses
GOLTOR 10 mg/10 mg
Kaltgeformte Durchdrückblister aus einer PVC/Aluminium-Folie/vinylbeschichtetes Aluminium versiegelt mit polyamidgedecktem Laminatfilm in Packungsgrößen zu 30, 100 Tabletten. Klinikpackungen zu 300 (10 x 30) Tabletten.
GOLTOR 10 mg/20 mg, GOLTOR 10 mg/40 mg, GOLTOR 10 mg/80 mg
Durchdrückblister aus opakem Polychlortrifluorethylen/PVC auf vinylbeschichtetem Aluminium in Packungen zu 30, 100 Tabletten.
Klinikpackungen zu 300 (10 x 30) Tabletten.
6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung
Keine besonderen Anforderungen.
7. INHABER DER ZULASSUNG
Merck Sharp & Dohme Limited Hertford Road, Hoddesdon Hertfordshire EN 11 9BU Vereinigtes Königreich
Mitvertrieb:
BERLIN-CHEMIE AG Glienicker Weg 125 12489 Berlin Deutschland
Telefon: (030) 6707-0 (Zentrale) Telefax: (030) 6707-2120 www.berlin-chemie .de
8. ZULASSUNGSNUMMERN
GOLTOR 10 mg/10 mg Tabletten: 58874.00.00 GOLTOR 10 mg/20 mg Tabletten: 58874.01.00 GOLTOR 10 mg/40 mg Tabletten: 58874.02.00 GOLTOR 10 mg/80 mg Tabletten: 58874.03.00
9. DATUM DER ERTEILUNG DER ZULASSUNG/VERLÄNGERUNG DER ZULASSUNG
Datum der Erteilung der Zulassung: 2. April 2004 Datum der Verlängerung der Zulassung: 17. Juni 2014
10. STAND DER INFORMATION
Februar 2016
11. VERKAUFSABGRENZUNG V erschreibungspflichtig.
GOLTOR-GPC-2016-02-03/Type II WS 262
33
Sofern nichts Gegenteiliges ausdrücklich betont oder aus dem Zusammenhang ersichtlich ist, gelten die nachfolgend für GOLTOR gemachten Ausführungen für alle Tablettenstärken.
Möglicherweise sind nicht alle Dosierungen in allen EU-Mitgliedstaaten auf dem Markt.