Haemate P 500
CSL Behring
Haemate® P 250/500/1000
1. BEZEICHNUNG DES ARZNEIMITTELS
Haemate® P 250/500/1000
Pulver und Lösungsmittel zur Herstellung einer Injektions- oder Infusionslösung.
2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
Eine Flasche enthält nominal:
- 250/500/1000 I.E. humanen Blutgerinnungsfaktor VIII (FVIII).
- 600/1200/2400 I.E. humanen von Willebrand Faktor (VWF).
Nach Rekonstitution mit 5 bzw. 10 ml enthält Haemate P 250/500 50 I.E./ml FVIII und 120 I.E./ml VWF. Nach Rekonstitution mit 15 ml enthält Haemate P 1000 66,6 I.E./ml FVIII und 160 I.E./ml VWF.
Die FVIII-Aktivität (I.E.) wird mittels chromogenem Test gemäß Europäischem Arzneibuch bestimmt. Die spezifische FVIII-Aktivität von Haemate beträgt etwa 2 - 6 I.E. FVIII/mg Protein.
Die VWF-Aktivität (I.E.) wird entsprechend der Ristocetin-Kofaktor-Aktivität (VWF:RCo) bestimmt, gemessen gegen den internationalen Standard für von Willebrand FaktorKonzentrate (WHO). Die spezifische VWF-Aktivität von Haemate beträgt etwa 5 - 17 I.E. VWF:RCo/mg Protein.
Haemate wird aus menschlichem Plasma hergestellt.
Sonstige Bestandteile mit bekannter Wirkung:
Natrium:
250/500 I.E. - etwa 113 mmol/l (2,6 mg/ml).
1000 I.E. - etwa 150 mmol/l (3,5 mg/ml).
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
3. DARREICHUNGSFORM
Pulver und Lösungsmittel zur Herstellung einer Injektions- oder Infusionslösung.
Weißes Pulver und klares, farbloses Lösungsmittel zur Herstellung einer Injektions- oder Infusionslösung.
4. KLINISCHE ANGABEN
4.1 Anwendungsgebiete
Von Willebrand Syndrom (VWS):
Prophylaxe und Therapie von Blutungen oder Blutungen während Operationen, wenn die Behandlung mit Desmopressin (DDAVP) alleine nicht wirksam oder kontraindiziert ist.
Hämophilie A (kongenitaler FVIII-Mangel):
Prophylaxe und Therapie von Blutungen bei Patienten mit Hämophilie A.
Dieses Produkt kann in der Behandlung des erworbenen Faktor-VIII-Mangels und zur Behandlung von Patienten mit Antikörpern gegen Faktor VIII eingesetzt werden.
4.2 Dosierung und Art der Anwendung
Die Therapie des von Willebrand Syndroms und der Hämophilie A soll unter der Aufsicht eines in der Hämophilie-Behandlung erfahrenen Arztes erfolgen.
Dosierung
Von Willebrand Syndrom:
Im Allgemeinen hebt 1 I.E./kg VWF:RCo den Plasmaspiegel des VWF:RCo um 0,02
I.E./ml
(2%) an.
Es sollten VWF:RCo Spiegel von > 0,6 I.E./ml (60%) und FVIII:C Spiegel von > 0,4 I.E./ml (40%) angestrebt werden.
Im Allgemeinen werden 40 - 80 I.E./kg von Willebrand Faktor (VWF:RCo) und 20 - 40 I.E. FVIII:C/kg Körpergewicht empfohlen, um eine Blutstillung zu erhalten.
Eine Anfangsdosis von 80 I.E./kg von Willebrand Faktor kann erforderlich sein, besonders bei Patienten mit Typ 3 von Willebrand Syndrom, bei denen zur Erhaltung adäquater Plasmaspiegel höhere Dosen als bei anderen Typen des von Willebrand Syndrom erforderlich sein können.
Vorbeugung von Blutungen im Fall von Operationen oder schweren Verletzungen:
Zur Vorbeugung von übermäßigen Blutungen während und nach Operationen sollte das Präparat 1 bis 2 Stunden vor der Operation verabreicht werden.
Eine angemessene Dosierung sollte alle 12 - 24 Stunden wiederholt werden. Dosierung und Dauer der Therapie richten sich nach dem klinischen Zustand des Patienten, nach Art und Ausmaß der Blutung und den VWF:RCo und FVIII:C Spiegeln.
Bei der Anwendung eines VWF Produktes sollte sich der behandelnde Arzt bewusst sein, dass eine kontinuierliche Behandlung einen übermäßigen Anstieg von FVIII:C verursachen kann. Zur Vermeidung eines unkontrollierten Anstiegs von FVIII:C nach einer 24 - 48 stündigen Behandlung sollte eine Reduzierung der Dosierung und/oder eine Verlängerung des Dosierungintervalls in Betracht gezogen werden.
Kinder und Jugendliche:
Die Dosierung bei Kindern richtet sich nach dem Körpergewicht und deshalb generell nach den gleichen Richtlinien wie für Erwachsene. Die Häufigkeit der Anwendung soll sich stets an der klinischen Wirksamkeit im Einzelfall orientieren.
Hämophilie A:
Zuvor unbehandelte Patienten:
Die Sicherheit und Wirksamkeit von Haemate bei zuvor unbehandelten Patienten wurde bisher nicht untersucht. Es liegen keine Daten vor.
Therapieüberwachung:
Zur Festlegung der benötigten Dosis und Infusionshäufigkeiten werden geeignete Bestimmungen der Faktor-VIII-Spiegel im Verlauf der Behandlung empfohlen. Das Ansprechen der jeweiligen Patienten auf Faktor VIII kann variieren, was sich an unterschiedlichen Halbwertszeiten und Recoveries zeigt. Die Dosierung auf Basis des Körpergewichts muss bei unter- oder übergewichtigen Patienten eventuell angepasst werden. Vor allem bei größeren chirurgischen Eingriffen ist eine genaue gerinnungsanalytische Überwachung (Faktor-VIII-Aktivität im Plasma) der Substitutionstherapie unerlässlich.
Die Patienten sollten bezüglich einer Entwicklung von Inhibitoren gegen Faktor VIII überwacht werden. Siehe auch Abschnitt 4.4.
Dosierung und Dauer der Substitutionstherapie richten sich nach dem Schweregrad des Faktor-VIII-Mangels, nach Ort und Ausmaß der Blutung und nach dem klinischen Zustand des Patienten.
Die Menge des zu verabreichenden Faktor VIII wird in Internationalen Einheiten (I.E.) angegeben, die dem derzeitigen WHO-Standard für Faktor-VIII-Produkte entsprechen. Die Faktor-VIII-Aktivität im Plasma wird entweder als Prozentsatz (bezogen auf das normale Humanplasma) oder bevorzugt in I.E. (bezogen auf den Internationalen Standard für Faktor VIII im Plasma) angegeben.
Eine I.E. Faktor-VIII-Aktivität entspricht dem Faktor-VIII-Gehalt von 1 ml normalem Humanplasma.
Bedarfsbehandlung:
Die Berechnung der benötigten Dosis an Faktor VIII basiert auf dem empirischen Ergebnis, dass 1 I.E. Faktor VIII pro kg Körpergewicht die Faktor-VIII-Aktivität im Plasma um ca.
2 % (2 I.E./dl) der normalen Aktivität anhebt. Die benötigte Dosis wird nach folgender Formel berechnet:
Erforderliche Einheiten = Körpergewicht (kg) x gewünschter Faktor-VIII-Anstieg (% oder I.E./dl) x 0,5.
Die Dosierung und Häufigkeit der Anwendung sollten sich stets an der klinischen Wirksamkeit im Einzelfall orientieren.
Bei den folgenden Blutungsereignissen soll die Faktor-VIII-Aktivität (in % der Norm oder I.E./dl) während des entsprechenden Zeitraums nicht unter den angegebenen Wert abfallen. Die folgende Tabelle dient als Empfehlung für die Dosierung bei Blutungsereignissen und chirurgischen Eingriffen:
|
Schweregrad der Blutung/Art des chirurgischen Eingriffs |
Benötigter Faktor-VIII-Spiegel (% oder I.E./dl) |
Häufigkeit der Dosierung (Stunden)/Dauer der Behandlung (Tage) |
|
Blutung | ||
|
Beginnende Hämarthrosen, Muskelblutungen oder Blutungen in der Mundhöhle |
20 - 40 |
Wiederholung der Infusion alle 12 bis 24 Stunden. Mind. 1 Tag, bis die Blutung (angezeigt durch Schmerz) gestillt oder bis die Wundheilung abgeschlossen ist. |
|
Ausgedehntere Hämarthrosen, Muskelblutungen oder Hämatome |
30 - 60 |
Wiederholung der Infusion alle 12 bis 24 Stunden für 3 bis 4 Tage, oder länger, bis zur Beseitigung des Schmerzzustandes und der akuten Bewegungseinschränkung. |
|
Lebensbedrohliche Blutungen |
60 - 100 |
Wiederholung der Infusion alle 8 bis 24 Stunden bis zur Aufhebung des lebensbedrohlichen Zustandes. |
|
Chirurgische Eingriffe | ||
|
Kleinere Eingriffe einschließlich Zahnextraktion |
30 - 60 |
Alle 24 Stunden, mind. 1 Tag, bis Abschluss der Wundheilung. |
|
Größere Eingriffe |
80 - 100 (prä- und |
Wiederholung der Infusion alle 8 bis 24 Stunden bis zum |
|
postoperativ) |
adäquaten Abschluss der Wundheilung, dann Behandlung für mind. weitere 7 Tage zur Erhaltung einer Faktor-VIII-Aktivität von 30 % bis 60 % | |
|
(I.E./dl). |
Prophylaxe:
Bei der Langzeitprophylaxe von Blutungen bei Patienten mit schwerer Hämophilie A betragen die üblichen Dosen 20 bis 40 I.E. Faktor VIII/kg Körpergewicht in Intervallen von 2 bis 3 Tagen. In manchen Fällen, besonders bei jungen Patienten, können kürzere Dosierungsintervalle oder höhere Dosen notwendig werden.
Kinder und Jugendliche:
Es liegen keine Daten aus klinischen Studien zur Dosierung von Haemate bei Kindern vor.
Art der Anwendung
Zur intravenösen Anwendung.
Das Produkt ist wie in Abschnitt 6.6 beschrieben zu lösen. Das gelöste Produkt soll vor der Anwendung auf Raum- oder Körpertemperatur angewärmt werden, und sollte langsam intravenös mit einer für den Patienten angenehmen Geschwindigkeit injiziert werden. Das Produkt sollte nach dem Überführen in die Spritze unverzüglich appliziert werden.
Falls eine größere Menge des Faktors substituiert werden soll, kann die Gabe auch als Infusion erfolgen. Überführen Sie dazu das gelöste Produkt in ein zugelassenes Infusionssystem.
Die Injektions- oder Infusionsgeschwindigkeit sollte nicht mehr als 4 ml pro Minute betragen. Der Patient ist auf jegliche Sofortreaktionen zu beobachten. Wenn eine Reaktion erfolgt, die mit der Anwendung von Haemate in Zusammenhang gebracht werden könnte, soll - in Abhängigkeit vom klinischen Zustand des Patienten - die Infusionsgeschwindigkeit gesenkt bzw. die Verabreichung abgebrochen werden (siehe auch Abschnitt 4.4).
4.3 Gegenanzeigen
Überempfindlichkeit gegen die Wirkstoffe oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Überempfindlichkeit
Allergische Überempfindlichkeitsreaktionen sind möglich. Falls Symptome einer Überempfindlichkeit auftreten, sollte den Patienten geraten werden, die Anwendung des Produkts sofort zu unterbrechen und ihren Arzt aufzusuchen. Die Patienten sollen über
Frühzeichen von Überempfindlichkeitsreaktionen informiert werden, wie z. B. quaddelartiger Hautausschlag, generalisierte Nesselsucht, Engegefühl in der Brust, pfeifendes Atemgeräusch, Hypotonie und Anaphylaxie.
Bei einem Schock sollen die aktuellen medizinischen Richtlinien zur Schockbehandlung beachtet werden.
Haemate enthält bis zu 70 mg Natrium pro 1000 I.E. Dies sollte bei Patienten, die eine salzarme Diät einhalten müssen, berücksichtigt werden.
Von Willebrand Syndrom
Das Auftreten thrombotischer Ereignisse inklusive Lungenembolien ist möglich, insbesondere bei Patienten mit bekannten Risikofaktoren (z. B. operative Eingriffe ohne Thromboseprophylaxe, mangelnde frühzeitige Mobilisierung, Übergewicht, Überdosierung, Krebs). Risikopatienten müssen auf Frühzeichen einer Thrombose beobachtet werden. Eine Prophylaxe gegen venöse Thromboembolien sollte entsprechend den aktuellen Empfehlungen eingeleitet werden.
Bei der Anwendung eines VWF Produktes sollte sich der behandelnde Arzt bewusst sein, dass eine kontinuierliche Behandlung einen übermäßigen Anstieg von FVIII:C verursachen kann. Bei Patienten, die FVIII-haltige VWF-Produkte erhalten, sollten die FVIII:C Plasmaspiegel überwacht werden, um länger anhaltende, übermäßig hohe FVIII:C Plasmaspiegel, die das Risiko von thrombotischen Ereignissen erhöhen, zu vermeiden. Antithrombotische Maßnahmen sollten dabei in Betracht gezogen werden.
Patienten mit VWS, besonders Typ 3 Patienten, können neutralisierende Antikörper (Inhibitoren) gegen den VWF entwickeln. Falls die erwarteten VWF:RCo Aktivitätsspiegel im Plasma nicht erreicht werden, oder falls die Blutung nicht mit einer geeigneten Dosis kontrolliert werden kann, sollte ein geeigneter Test durchgeführt werden, um das Vorliegen von VWF Inhibitoren zu bestimmen. Bei Patienten mit hohen Inhibitorspiegeln ist die Möglichkeit eines Therapieversagens gegeben, und andere therapeutische Maßnahmen sollten in Betracht gezogen werden.
Hämophilie A
Inhibitoren:
Die Bildung neutralisierender Antikörper (Inhibitoren) gegen Faktor VIII ist eine bekannte Komplikation bei der Behandlung von Patienten mit Hämophilie A. Diese Inhibitoren sind gewöhnlich IgG Immunglobuline, die sich gegen die Faktor-VIII-Gerinnungsaktivität richten. Sie werden mittels modifiziertem Test in Bethesda-Einheiten (BE) pro ml Plasma quantifiziert. Das Risiko der Bildung von Inhibitoren korreliert mit der Exposition gegenüber antihämophilem Faktor VIII, wobei das Risiko in den ersten 20 Expositionstagen am höchsten ist. In seltenen Fällen entwickeln sich Inhibitoren noch nach den ersten 100 Expositionstagen.
Fälle von wiederkehrenden Inhibitoren (mit niedrigem Inhibitortiter/Inhibitorspiegel) wurden nach Umstellen von einem FVIII-Produkt zu einem anderen bei zuvor behandelten Patienten mit mehr als 100 Expositionstagen beobachtet, die eine Inhibitorentwicklung in der Vorgeschichte haben. Deshalb wird empfohlen, alle Patienten nach jedem Wechsel des Produktes sorgfältig auf das Auftreten von Inhibitoren zu beobachten.
Grundsätzlich sollten alle Patienten, die mit humanem Gerinnungsfaktor VIII behandelt wurden, durch geeignete klinische Beobachtung und Labortests sorgfältig bezüglich der Entwicklung von Inhibitoren untersucht werden. Wenn der erwartete Plasmaspiegel der FVIII-Aktivität im Plasma nicht erreicht wird, oder wenn die Blutung nicht mit einer angemessenen Dosis kontrolliert wird, soll ein Test zum Nachweis von FVIII-Inhibitoren durchgeführt werden. Bei Patienten mit hohen Inhibitorspiegeln kann die FVIII-Behandlung unwirksam sein und es sollten andere Behandlungsmöglichkeiten erwogen werden. Die Behandlung solcher Patienten soll von Ärzten, die in der Therapie von Hämophilie A-Patienten und Patienten mit FVIII-Inhibitoren erfahren sind, durchgeführt werden. Siehe auch Abschnitt 4.8 „Nebenwirkungen“.
Kardiovaskuläre Ereignisse:
Bei Patienten mit bestehenden kardiovaskulären Risikofaktoren, kann eine Substitutionstherapie mit FVIII das kardiovaskuläre Risiko erhöhen.
Katheter-assoziierte Komplikationen:
Wenn ein zentralvenöser Katheter (ZVK) erforderlich ist, sollte das Risiko von Katheterassoziierten Komplikationen einschließlich lokaler Infektionen, Bakteriämie und Katheterassoziierten Thrombosen berücksichtigt werden.
Virussicherheit
Standardmethoden zur Vermeidung von Infektionskrankheiten, die im Rahmen der Anwendung von aus menschlichem Blut oder Plasma hergestellten Arzneimitteln auftreten können, umfassen die Auswahl der Spender, die Prüfung jeder einzelnen Spende und jedes Plasmapools auf spezifische Marker für Infektionen sowie die Einbeziehung effektiver Herstellungsschritte zur Inaktivierung/Eliminierung von Viren. Trotz dieser Maßnahmen kann die Möglichkeit der Übertragung von Erregern bei der Anwendung von aus menschlichem Blut oder Plasma hergestellten Arzneimitteln nicht vollständig ausgeschlossen werden. Dies gilt auch für bisher unbekannte Viren und andere Pathogene.
Die getroffenen Maßnahmen werden als wirksam angesehen für umhüllte Viren, wie z. B. das humane Immundefizienzvirus (HIV), das Hepatitis B-Virus (HBV) und das Hepatitis C-Virus (HCV) sowie für das nicht umhüllte Hepatitis A-Virus.
Für andere, nicht-umhüllte Viren, wie z. B. Parvovirus B19, können die getroffenen Maßnahmen von eingeschränktem Wert sein.
Parvovirus B19 Infektionen können schwerwiegende Folgen für schwangere Frauen (fetale Infektion) und für Personen mit Immunmangelkrankheiten oder gesteigerter Erythropoese (z.B. hämolytische Anämie) haben.
Für Patienten, die regelmäßig Blutgerinnungsfaktor VIII oder von Willebrand FaktorPräparate aus menschlichem Blut oder Plasma erhalten, wird eine Impfung gegen Hepatitis A und Hepatitis B empfohlen.
Es wird auf die Dokumentationspflicht gemäß Transfusionsgesetz hingewiesen.
4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Es sind bisher keine Wechselwirkungen von VWF und FVIII mit anderen Arzneimitteln untersucht worden.
4.6 Fertilität, Schwangerschaft und Stillzeit
Reproduktionsstudien am Tier wurden mit Haemate nicht durchgeführt.
Von Willebrand Syndrom
Beim von Willebrand Syndrom sind Frauen wegen der autosomalen Vererbung und aufgrund ihrer zusätzlichen Blutungsrisiken wie Menstruation, Schwangerschaft, Wehen, Geburt sowie gynäkologischer Beschwerden mehr als Männer betroffen. Aufgrund der allgemeinen Erfahrungen kann die VWF Substitution zur Vorbeugung oder Therapie von akuten Blutungen empfohlen werden. Klinische Studien zur Substitutionstherapie mit VWF in der Schwangerschaft und Stillzeit liegen jedoch nicht vor.
Hämophilie A
Aufgrund des seltenen Vorkommens der Hämophilie A bei Frauen liegen keine Erfahrungen über die Anwendung von FVIII während der Schwangerschaft und Stillzeit vor.
Daher sollen VWF und FVIII in der Schwangerschaft und Stillzeit nur bei klarer Indikationsstellung angewendet werden.
4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Haemate hat keinen Einfluss auf die Verkehrstüchtigkeit und Fähigkeit zum Bedienen von Maschinen.
4.8 Nebenwirkungen
Die folgenden Informationen zu Unverträglichkeitsreaktionen basieren auf der Erfahrung von Spontanmeldungen.
Zusammenfassung des Sicherheitsprofiles
Während der Behandlung mit Haemate bei Erwachsenen und Heranwachsenden können die folgenden Nebenwirkungen auftreten: Überempfindlichkeitsreaktionen oder allergische Reaktionen, thromboembolische Ereignisse und Fieber. Desweiteren können Patienten Inhibitoren gegen FVIII und VWF entwickeln.
Tabellarische Auflistung der Nebenwirkungen
Die unten aufgeführte Tabelle entspricht der MedDRA Systemorganklassifikation.
Die Häufigkeiten wurden entsprechend der nachfolgenden Konventionen berechnet: sehr häufig (>1/10); häufig (> 1/100 und < 1/10); gelegentlich (> 1/1000 und < 1/100); selten (> 1/10.000 und < 1/1000); sehr selten (< 1/10.000); nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar)
|
MedDRA Systemorganklasse |
Nebenwirkung |
Häufigkeit |
|
Erkrankungen des Blutes und des Lymphsystems |
Hypervolämie Hämolyse VWF Inhibitoren FVIII Inhibitoren |
Nicht bekannt Nicht bekannt Sehr selten Sehr selten |
|
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort |
Fieber |
Sehr selten |
|
Erkrankungen des Immunsystems |
Überempfindlichkeitsreaktionen (allergische Reaktionen) |
Sehr selten |
|
Gefäßerkrankungen |
Thrombosen Thromboembolien |
Sehr selten Sehr selten |
Beschreibung ausgewählter Nebenwirkungen
• Erkrankungen des Blutes und des Lymphsystems
Wenn sehr hohe oder häufig wiederholte Dosen benötigt werden, wenn Inhibitoren vorliegen oder wenn es sich um prä- oder postoperative Behandlung handelt, sollten die Patienten sorgfältig auf Hinweise einer Hypervolämie beobachtet werden. Zusätzlich sollten solche Patienten mit Blutgruppen A, B und AB auf Anzeichen von intravaskulärer Hämolyse und/oder abnehmende Hämatokritwerte beobachtet werden.
• Allgemeine Erkrankungen und Beschwerden am Verabreichungsort In sehr seltenen Fällen wurde Fieber beobachtet.
• Erkrankungen des Immunsystems
Überempfindlichkeitsreaktionen oder allergische Reaktionen (die auch Angioödem,
Brennen und Stechen an der Infusionsstelle, Schüttelfrost, Hautrötung mit Hitzegefühl, generalisierte Urtikaria, Kopfschmerzen, Nesselausschlag, Hypotonie, Lethargie, Übelkeit, Unruhe, Tachykardie, Engegefühl in der Brust, Kribbeln, Erbrechen und Stridor mit einschließen können) wurden mit sehr seltener Häufigkeit beobachtet und können sich in manchen Fällen zu schwerer Anaphylaxie (einschließlich Schock) entwickeln.
Von Willebrand Syndrom
• Erkrankungen des Blutes und des Lymphsystems
Patienten mit VWS, besonders Typ 3 Patienten, können sehr selten neutralisierende Antikörper (Inhibitoren) gegen den VWF entwickeln. Wenn solche Inhibitoren auftreten, manifestiert sich der Zustand als unzureichende klinische Antwort. Da es sich um präzipitierende Antikörper handelt, können sie zeitgleich mit anaphylaktischen Reaktionen auftreten. Deshalb sollten Patienten mit anaphylaktischen Reaktionen auf die Anwesenheit von Inhibitoren getestet werden.
In solchen Fällen wird empfohlen, ein spezialisiertes Hämophilie-Zentrum aufzusuchen.
• Gefäßerkrankungen
Sehr selten können Thrombosen und/oder Thromboembolien (inklusive Lungenembolien) auftreten.
Bei Patienten, die VWF Produkte erhalten, können länger anhaltende, übermäßig hohe FVIII:C Plasmaspiegel das Risiko von thrombotischen Ereignissen erhöhen (siehe auch Abschnitt 4.4).
Hämophilie A
Erkrankungen des Blutes und des Lymphsystems
Patienten mit Hämophilie A können sehr selten neutralisierende Antikörper (Inhibitoren) gegen Faktor VIII entwickeln. Wenn solche Inhibitoren auftreten, manifestiert sich der Zustand als unzureichende klinische Antwort. In solchen Fällen wird empfohlen, ein spezialisiertes Hämophilie-Zentrum aufzusuchen.
Informationen zur Virussicherheit siehe Abschnitt 4.4.
Kinder und Jugendliche
Es wird erwartet, dass die Häufigkeit, Art und Schwere der Nebenwirkungen bei Kindern denen bei Erwachsenen entspricht.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-RisikoVerhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel, Paul-Ehrlich-Institut, Paul-Ehrlich-Str. 51-59, 63225 Langen, Tel: +49 6103 77 0, Fax: +49 6103 77 1234, Website: www.pei.de anzuzeigen.
4.9 Überdosierung
Symptome einer Überdosierung mit VWF und FVIII sind bisher nicht berichtet. Dennoch kann das Risiko einer Thrombose bei einer äußerst hohen Dosis nicht ausgeschlossen werden, besonders bei FVIII-haltigen VWF-Produkten mit einem hohen FVIII Gehalt.
5. PHARMAKOLOGISCHE EIGENSCHAFTEN
5.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Antihämorrhagika: Blutgerinnungsfaktoren,
Von Willebrand Faktor und Blutgerinnungsfaktor VIII in Kombination.
ATC-Code: B02BD06
Von Willebrand Faktor
Haemate verhält sich genauso wie der körpereigene VWF.
Zusätzlich zu seiner Funktion als Faktor VIII stabilisierendes Protein vermittelt von Willebrand Faktor die Anlagerung der Thrombozyten an den verletzten Gefäßendothelien und spielt die Hauptrolle bei der Thrombozytenaggregation.
Bei Patienten mit VWF-Mangel (VWS) führt die Verabreichung des VWF zu einer Korrektur der Blutgerinnungsstörungen in 2 Stufen:
- VWF vermittelt die Bindung der Plättchen an das Subendothelgewebe verletzter Gefäße (VWF bindet sowohl an Gefäßsubendothelgewebe als auch an die Plättchenmembran) und führt somit zu einer primären Blutstillung, was an der Verkürzung der Blutungszeit erkennbar ist. Dieser Effekt tritt unmittelbar ein und ist vorrangig abhängig vom hohen Anteil an hochmolekularen VWF-Multimeren.
- VWF führt zeitverzögert zu einer Korrektur des assoziierten FVIII-Mangels. Nach intravenöser Gabe bindet VWF an endogenen FVIII (der physiologischerweise vom Patienten selbst gebildet wird), stabilisiert diesen Faktor und verhindert somit dessen schnellen Abbau.
Deshalb bewirkt die Gabe von reinem VWF (VWF Präparat mit einem niedrigen FVIII Gehalt) nach der ersten Infusion als Nebeneffekt mit einer gewissen Verzögerung die Wiederherstellung des normalen Faktor VIII-Spiegels.
Die Gabe von FVIII-haltigen VWF-Präparaten bringt den FVIII:C Spiegel sofort nach der ersten Infusion auf ein normales Niveau.
Faktor VIII
Haemate verhält sich genauso wie der körpereigene Faktor VIII.
Der Faktor-VIII-/von Willebrand Faktor-Komplex besteht aus 2 Proteinen (Faktor VIII und von Willebrand Faktor), die unterschiedliche physiologische Wirkungen besitzen.
Bei der Behandlung hämophiler Patienten, bindet Faktor VIII an von Willebrand Faktor im Blutkreislauf des Patienten.
Aktivierter Faktor VIII agiert als Cofaktor für den aktivierten Faktor IX und beschleunigt so die Umwandlung von Faktor X in aktivierten Faktor X. Aktivierter Faktor X wiederum wandelt Prothrombin in Thrombin um. Thrombin wandelt dann Fibrinogen in Fibrin um, und ein Blutgerinnsel kann gebildet werden. Hämophilie A ist eine geschlechtsgebundene vererbbare Störung der Blutgerinnung, die auf einem Mangel an Faktor VIII beruht. Als Folge davon kann es zu starken Blutungen in Gelenke, Muskeln oder innere Organe kommen, die entweder spontan oder infolge von Unfällen oder chirurgischen Eingriffen entstehen können. Durch Substitutionstherapie werden die Faktor-VIII-Plasmaspiegel angehoben und dadurch eine vorübergehende Korrektur des Faktor-Mangels sowie eine Behebung der Blutungsneigung herbeigeführt.
5.2 Pharmakokinetische Eigenschaften
Von Willebrand Faktor
Die Pharmakokinetik von Haemate wurde bei 28 Patienten mit VWS (Typ 1 n=10; Typ 2A n=10; Typ 2M n=1; Typ 3 n=7) im nicht blutenden Stadium untersucht. Die mediane terminale Halbwertszeit von VWF:RCo (2-Kompartment-Modell) war 9,9 Stunden (Bereich
2.8 bis 51,1 Stunden). Die mediane initiale Halbwertszeit beträgt 1,47 Stunden (Bereich
0. 28 bis 13,86 Stunden). Die mediane in vivo Recovery für die VWF:RCo Aktivität betrug
1.9 (I.E./dl)/(I.E./kg) [Bereich 0,6 bis 4,5 (I.E./dl)/(I.E./kg)]. Die mediane AUC betrug 1664
1. E./dl*h (Bereich 142 bis 3846 I.E./dl*h), die mediane MRT war 13,7 Stunden (Bereich 3,0 bis 44,6 Stunden) und die mediane Clearance war 4,81 ml/kg/h (Bereich 2,08 bis 53,0 ml/kg/h).
Die höchsten Plasmaspiegel für VWF sind ungefähr 50 Minuten nach der Injektion messbar. Die höchsten FVIII Spiegel treten zwischen 1 und 1,5 Stunden nach der Injektion auf.
Faktor VIII
Nach intravenöser Gabe steigt die Faktor-VIII-Aktivität (FVIII:C) schnell an, gefolgt von einer zunächst schnellen und einer nachfolgend langsameren Phase der Aktivitätsabnahme. Studien bei Patienten mit Hämophilie A zeigten eine mediane Halbwertszeit von 12,6 Stunden (Bereich 5,0 bis 27,7 Stunden). Es wurde eine allgemeine mediane FVIII in vivo Recovery von 1,73 I.E./dl pro I.E./kg (Bereich 0,5 bis 4,13) erreicht. In einer Studie war die mediane Verweildauer (MRT) 19,0 Stunden (Bereich 14,8 bis 40,0 Stunden), die mediane Fläche unter der Kurve (AUC) war 36,1 (% * Stunden) / (I.E./kg) (Bereich 14,8 bis 72,4 (% * Stunden) / (I.E./kg)), die mediane Clearance 2,8 ml/h/kg (Bereich 1,4 bis 6,7 ml/h/kg).
Kinder und Jugendliche
Es liegen keine pharmakokinetischen Daten für Kinder unter 12 Jahren vor.
5.3 Präklinische Daten zur Sicherheit
Die in Haemate enthaltenen arzneilich wirksamen Bestandteile FVIII und von Willebrand Faktor werden aus humanem Plasma gewonnen und verhalten sich wie körpereigene Plasmabestandteile. Die einmalige Verabreichung von Haemate an verschiedenen Tierspezies gab keine Hinweise auf toxische Auswirkungen. Präklinische Studien mit wiederholten Dosisgaben (chronische Toxizität, Kanzerogenität, Reproduktionstoxizität) können in herkömmlichen Tiermodellen nicht sinnvoll durchgeführt werden, da aufgrund der Verabreichung heterologer humaner Proteine Antikörper gebildet werden.
6. PHARMAZEUTISCHE ANGABEN
6.1 Liste der sonstigen Bestandteile
Human-Albumin, Glycin, Natriumchlorid, Natriumcitrat, NaOH und HCl (in geringen Mengen) zur Einstellung des pH-Wertes.
Beigefügtes Lösungsmittel: Wasser für Injektionszwecke 5/10/15 ml
6.2 Inkompatibilitäten
Dieses Arzneimittel darf, außer mit den unter Abschnitt 6.1 aufgeführten Lösungsmitteln, nicht mit anderen Arzneimitteln, Lösungs- oder Verdünnungsmitteln vermischt werden.
6.3 Dauer der Haltbarkeit
3 Jahre.
Nach Rekonstitution ist die physiko-chemische Stabilität für 48 Stunden bei Raumtemperatur (bis max. +25 °C) belegt. Aus mikrobiologischer Sicht und da Haemate kein Konservierungsmittel enthält, sollte das gelöste Präparat sofort verbraucht werden. Falls es nicht sofort angewendet wird, soll eine Aufbewahrung 8 Stunden bei Raumtemperatur nicht überschreiten.
Das Produkt sollte nach dem Überführen in die Spritze unverzüglich appliziert werden.
6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Haemate nicht über +25 °C lagern.
Nicht einfrieren. In der geschlossenen Faltschachtel aufbewahren.
6.5 Art und Inhalt des Behältnisses
Eine Packung mit 250 I.E. enthält:
1 Vakuumflasche mit Pulver 1 Flasche mit 5 ml Wasser für Injektionszwecke Eine Packung mit Zubehör enthält:
1 Filter Transfer Set 20/20 1 Einmalspritze (5 ml)
1 Venenpunktionsbesteck
2 Alkoholtupfer 1 Pflaster
Eine Packung mit 500 I.E. enthält:
1 Vakuumflasche mit Pulver 1 Flasche mit 10 ml Wasser für Injektionszwecke Eine Packung mit Zubehör enthält:
1 Filter Transfer Set 20/20 1 Einmalspritze (10 ml)
1 Venenpunktionsbesteck
2 Alkoholtupfer 1 Pflaster
Eine Packung mit 1000 I.E. enthält 1 Vakuumflasche mit Pulver 1 Flasche mit 15 ml Wasser für Injektionszwecke Eine Packung mit Zubehör enthält:
1 Filter Transfer Set 20/20 1 Einmalspritze (20 ml)
1 Venenpunktionsbesteck
2 Alkoholtupfer 1 Pflaster
6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung
Art der Anwendung
Allgemeine Hinweise
- Die Lösung sollte klar oder leicht opaleszent sein. Rekonstituiertes Produkt sollte nach der Filtration/Aufziehen der Lösung in die Spritze (siehe unten) und vor der Anwendung auf Partikel und Verfärbungen visuell überprüft werden. Auch wenn die Hinweise zur Rekonstitution sorgfältig befolgt wurden, kann die Lösung einige Flocken oder Partikel enthalten, die jedoch durch den im Mix2Vial enthaltenen Filter vollständig zurückgehalten werden. Die errechnete Dosierung wird durch das Filtrieren nicht beeinflusst. Deutlich trübe Lösungen und Lösungen mit Ausflockungen oder Niederschlag nach Filtration dürfen nicht verwendet werden.
- Zubereitung und Entnahme müssen unter aseptischen Bedingungen erfolgen. Zubereitung
Erwärmen Sie das Lösungsmittel auf Raumtemperatur. Vor dem Öffnen der Mix2Vial Packung die Flip-Off-Kappen der Lösungsmittel- und Produktflaschen entfernen und die Stopfen mit einer antiseptischen Lösung behandeln und anschließend trocknen lassen.
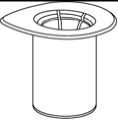
1
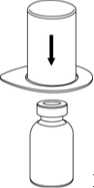
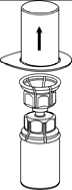
3
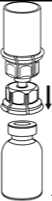
4
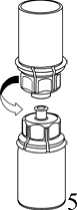
1. Entfernen Sie das Deckpapier von der Mix2Vial Packung. Das Mix2Vial nicht aus dem Blister entnehmen!
2. Die Lösungsmittelflasche auf eine ebene, saubere Fläche stellen und festhalten. Das Mix2Vial Set mit dem Blister greifen und den Dorn des blauen Adapters senkrecht in den Stopfen der Lösungsmittelflasche einstechen.
3. Vorsichtig die Verpackung vom Mix2Vial Set entfernen, indem man den Blister am Siegelrand fasst und ihn senkrecht nach oben abzieht. Dabei ist darauf zu achten, dass nur der Blister und nicht das Mix2Vial entfernt wird.
4. Die Produktflasche auf eine feste Unterlage stellen. Die Lösungsmittelflasche mit dem aufgesetzten Mix2Vial Set herumdrehen und den Dorn des transparenten Adapters senkrecht in den Stopfen der Produktflasche einstechen. Das Lösungsmittel läuft automatisch in die Produktflasche über.
5. Mit der einen Hand die Produktseite und mit der anderen Hand die Lösungsmittelseite des Mix2Vial greifen und das Set vorsichtig auseinander schrauben, um eine übermäßige Schaumbildung beim Auflösen des Produktes zu vermeiden. Entsorgen Sie die Lösungsmittelflasche mit dem blauen Mix2Vial Adapter.
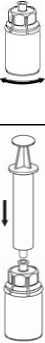
6. Die Produktflasche mit dem transparenten Adapter vorsichtig schwenken, bis das Produkt vollständig gelöst ist. Nicht schütteln.
6__
7. Luft in eine leere, sterile Spritze aufziehen. Die Produktflasche aufrecht halten, die Spritze mit dem Luer Lock Anschluss des Mix2Vial Set verbinden und die Luft in die Produktflasche injizieren.
Aufziehen der Lösung in die Spritze und Anwendung
|
i |
*8 |
8. Den Stempel der Spritze gedrückt halten, das gesamte System herumdrehen und das Produkt durch langsames Zurückziehen der Kolbenstange in die Spritze aufziehen. |
|
1 |
9. Nachdem die Lösung vollständig in die Spritze überführt ist, den Spritzenzylinder fassen (dabei die Kolbenstange in ihrer Position halten) und die Spritze vom transparenten Mix2Vial-Adapter abdrehen. |
Zur Injektion von Haemate werden Kunststoff-Einmalspritzen empfohlen, da geschliffene Oberflächen von Glasspritzen bei derartigen Lösungen zur Verklebung neigen.
Die Lösung langsam intravenös verabreichen (siehe Abschnitt 4.2). Sorgfältig darauf achten, dass kein Blut in die mit Produkt gefüllte Spritze gelangt.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den lokalen Anforderungen zu entsorgen.
7. INHABER DER ZULASSUNG
CSL Behring GmbH
- Emil-von-Behring-Str. 76 35041 Marburg
- Verkauf Deutschland Philipp-Reis-Str. 2 65795 Hattersheim Tel.: (069) 305 - 8 44 37 Fax: (069) 305 - 1 71 29
8. ZULASSUNGSNUMMERN
Haemate® P 250 Zul.-Nr.: 2416.00.00
Haemate® P 500 Zul.-Nr.: 2416.01.00
Haemate® P 1000 Zul.-Nr.: 2416.02.00
9. DATUM DER ERTEILUNG DER ZULASSUNG/VERLÄNGERUNG DER
ZULASSUNG
Erstzulassung: 07. Juni 1982
Letzte Verlängerung der Zulassung: 09. Juni 2002
10. STAND DER INFORMATION Oktober 2016
11. HERKUNFTSLÄNDER DES BLUTPLASMAS
Belgien, Deutschland, Österreich, Polen, Ungarn, USA
12. VERSCHREIBUNGSSTATUS
Verschreibungspflichtig