Haemoctin Sdh 250
1. BEZEICHNUNG DES ARZNEIMITTELS
Haemoctin SDH 250 Haemoctin SDH 500 Haemoctin SDH 1000
Pulver und Lösungsmittel zur Herstellung einer Injektionslösung Aus Plasma vom Menschen gewonnener Blutgerinnungsfaktor VIII
2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
Eine Durchstechflasche enthält nominell 250, 500 bzw. 1000 I.E. aus Plasma vom Menschen gewonnenen Blutgerinnungsfaktor VIII.
Haemoctin SDH 250 bzw. Haemoctin SDH 500 enthält nach Lösen in 5 ml bzw. 10 ml Wasser für Injektionszwecke ca. 50 I.E./ml Blutgerinnungsfaktor VIII vom Menschen. Haemoctin SDH 1000 enthält nach Lösen in 10 ml Wasser für Injektionszwecke ca. 100 I.E./ml Blutgerinnungsfaktor VIII vom Menschen.
Zur Bestimmung der Stärke (I.E.) wird der chromogene Faktor-VIII-Gerinnungstest gemäß Europäischem Arzneibuch verwendet; die spezifische Aktivität von Haemoctin SDH 250, 500 bzw. 1000 beträgt ca. 100 I.E./mg Protein.
Hergestellt aus Plasma menschlicher Spender.
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
3. DARREICHUNGSFORM
Pulver und Lösungsmittel zur Herstellung einer Injektionslösung.
Weißes Pulver und klares, farbloses Lösungsmittel zur Herstellung einer Injektionslösung
4. KLINISCHE ANGABEN
4.1 Anwendungsgebiete
Therapie und Prophylaxe von Blutungen bei Patienten mit Hämophilie A (angeborener Faktor-VIII-Mangel).
Dieses Produkt enthält den von-Willebrand-Faktor nicht in pharmakologisch wirksamer Menge und ist daher nicht für die Behandlung der von-Willebrand-Krankheit indiziert.
4.2 Dosierung und Art der Anwendung
Die Behandlung sollte unter Überwachung eines Arztes erfolgen, der mit der Therapie von Hämophilie vertraut ist.
Dosierung
Die Dosierung und Dauer der Substitutionstherapie sind abhängig von der Schwere des Faktor-VIII-Mangels sowie von Lokalisation und Ausmaß der Blutung und vom klinischen Zustand des Patienten.
Die verabreichten Faktor-VIII-Einheiten werden in Internationalen Einheiten (I.E.) angegeben, abgeleitet vom aktuellen WHO-Standard für Faktor-VIII-Produkte. Die Faktor-VIII-Aktivität im Plasma wird entweder als Prozentsatz (bezogen auf normales Humanplasma) oder in
Internationalen Einheiten (bezogen auf den Internationalen Standard für Faktor VIII im Plasma) angegeben.
Eine Internationale Einheit (I.E.) Faktor VIII-Aktivität entspricht der Menge an Faktor-VIII in einem Milliliter normalen menschlichem Plasma. Die Berechnung der erforderlichen Faktor-VIII-Dosierung basiert auf dem empirischen Befund, dass die Gabe von 1 Internationalen Einheit (I.E.) Faktor VIII pro kg Körpergewicht die Faktor-VIII-Aktivität im Plasma um 1 %-2 %, bezogen auf den Normalwert, anhebt.
Die erforderliche Dosis wird mit der folgenden Formel berechnet:
Benötigte Einheiten = Körpergewicht (kg) x gewünschter Faktor-VIII-Anstieg (%) x 0,5
Die Dosis und die Häufigkeit der Verabreichung sollten sich immer an der klinischen Wirksamkeit im jeweiligen Einzelfall orientieren.
Im Fall der aufgeführten Blutungsereignisse sollte die Faktor-VIII-Aktivität im entsprechenden Zeitraum nicht unter das angegebene Niveau (in % der Norm) fallen. Die folgende Tabelle kann als Richtlinie für die Dosierung bei Blutungsereignissen und chirurgischen Eingriffen dienen:
|
Schwere der Blutung/Art des chirurgischen Eingriffs |
Benötigter Faktor-VIII-Plasma-Spiegel (%) |
Häufigkeit der Dosierung (Stunden)/ Behandlungsdauer (Tage) |
|
Blutungen | ||
|
Gelenkblutungen im Frühstadium, Muskelblutungen, Blutungen im Mundbereich |
20 - 40 |
Injektion alle 12 bis 24 Stunden; mindestens 1 Tag, bis die (durch Schmerzen erkennbare) Blutung sistiert bzw. Wundheilung erreicht ist. |
|
Ausgeprägtere Gelenkblutungen, Muskelblutungen oder Hämatome |
30 - 60 |
Injektion alle 12 bis 24 Stunden für 3 bis 4 Tage oder länger wiederholen, bis die Schmerzen und akute Behinderungen beseitigt sind. |
|
Lebensbedrohliche Blutungen |
60 - 100 |
Injektion alle 8 bis 24 Stunden wiederholen, bis die Gefahr vorüber ist. |
|
Chirurgische Eingriffe | ||
|
Kleinere Eingriffe einschließlich Zahnextraktionen |
30 - 60 |
Injektion alle 24 Stunden; mindestens 1 Tag, bis die Wundheilung erreicht ist. |
|
Größere Eingriffe |
80 - 100 (prä- und postoperativ) |
Injektion alle 8 bis 24 Stunden wiederholen, bis ausreichende Wundheilung erreicht ist; dann für mindestens weitere 7 Tage einen Faktor-VIII-Spiegel von 30 % bis 60 % aufrechterhalten. |
Prophylaxe
Bei der Langzeitprophylaxe von Blutungen bei Patienten mit schwerer Hämophilie A beträgt die übliche Dosis 20 bis 40 I.E. Faktor VIII pro kg Körpergewicht im Abstand von 2-3 Tagen. In manchen Fällen, insbesondere bei jüngeren Patienten, können kürzere Dosierungsintervalle oder höhere Dosen erforderlich sein.
Während des Behandlungsverlaufs wird, zur Steuerung der zu verabreichenden Dosis und der Häufigkeit der Injektionen, eine angemessene Bestimmung der Faktor-VIII-Plasmaspiegel angeraten. Besonders bei größeren chirurgischen Eingriffen ist eine genaue Überwachung der
Substitutionstherapie durch Bestimmung des Blutgerinnungsstatus (Faktor-VIII-Aktivität) unerlässlich. Einzelne Patienten können sich in ihrer Reaktion auf Faktor VIII unterscheiden, verschiedene in vivo Wiederfindungsraten erreichen und unterschiedliche Halbwertszeiten aufweisen.
Patienten sollten auf die Bildung von Hemmkörpern gegen Faktor VIII überwacht werden. Falls die erwarteten Faktor-VIII-Aktivitäten im Plasma nicht erreicht werden oder die Blutung mit einer angemessenen Dosis nicht beherrscht wird, muss ein Hemmkörpertest durchgeführt werden. Bei Patienten mit hohen Hemmkörperspiegeln ist die Faktor-VIII-Therapie möglicherweise nicht wirksam und es sollten therapeutische Alternativen in Erwägung gezogen werden.
Diese Patienten sollten nur unter Aufsicht eines Arztes behandelt werden, der Erfahrung mit der Behandlung von Hämophilie-Patienten mit Faktor-VIII-Inhibitoren hat. Siehe auch Abschnitt 4.4.
Nicht vorbehandelte Patienten Es liegen keine Daten vor.
Art der Anwendung
Das Produkt wird intravenös verabreicht. Es wird empfohlen, die maximale Infusionsrate von Haemoctin SDH 250, 500 bzw. 1000 (2 - 3 ml/min) nicht zu überschreiten. Hinweise zur Rekonstitution des Arzneimittels vor der Anwendung, siehe Abschnitt 6.6.
4.3 Gegenanzeigen
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Überempfindlichkeit
Wie bei jedem intravenös zu verabreichenden proteinhaltigen Produkt sind allergische Überempfindlichkeitsreaktionen möglich. Das Produkt enthält Spuren anderer humaner Proteine außer Faktor VIII. Die Patienten sind darüber zu informieren, dass die Behandlung sofort abgebrochen und ein Arzt konsultiert werden muss, wenn Symptome einer Überempfindlichkeitsreaktion auftreten. Patienten sollen über das mögliche Auftreten von frühen Zeichen einer Überempfindlichkeitsreaktion, wie z. B. Ausschlag, generalisierte Urtikaria, Brustenge, Stridor, Hypotonie und Anaphylaxis informiert werden.
Beim Auftreten eines Schocks ist die medizinische Standardtherapie bei Schock durchzuführen.
Hemmkörper (Inhibitoren)
Die Bildung von neutralisierenden Antikörpern (Hemmkörpern) gegen Faktor VIII ist eine bekannte Komplikation der Hämophilie-A-Therapie. Diese Hemmkörper sind üblicherweise IgG-Immunglobuline, die die gerinnungsfördernde Aktivität des Faktor VIII beeinträchtigen.
Sie werden mit einem modifizierten Testverfahren bestimmt und in Bethesda-Einheiten (BE) pro Mililiter Plasma angegeben. Das Risiko der Bildung von Hemmkörpern korreliert mit der Exposition gegenüber Faktor VIII und ist innerhalb der ersten 20 Expositionstage am höchsten. Die Bildung von Hemmkörpern nach den ersten 100 Expositionstagen ist selten.
Fälle rezidivierender Hemmkörper (niedrige Titer) wurden nach Wechsel von einem Faktor-VIII-Präparat auf ein anderes bei vorbehandelten Patienten mit mehr als 100 Expositionstagen, die zuvor Hemmkörper gebildet hatten, beobachtet. Daher wird empfohlen, nach einem Präparate-Wechsel alle Patienten sorgfältig auf das Auftreten von Hemmkörpern zu überwachen.
Allgemein sollten alle mit humanen Blutgerinnungsfaktor VIII Präparaten behandelte Patienten mittels geeigneter klinischer Untersuchungen und Labortests sorgfältig auf die Bildung von Hemmkörpern überwacht werden. Falls die erwarteten Faktor-VIII-Aktivitäten im Plasma nicht erreicht werden oder die Blutung mit einer angemessenen Dosis nicht beherrscht wird, muss ein Hemmkörpertest durchgeführt werden. Bei Patienten mit hohen Hemmkörperspiegeln ist die Faktor-VIII-Therapie möglicherweise nicht wirksam und es sollten therapeutische Alternativen in Erwägung gezogen werden. Diese Patienten sollten nur unter Aufsicht eines Arztes behandelt werden, der Erfahrung mit der Behandlung von Hämophilie-Patienten mit Faktor-VIII-Inhibitoren hat.
Katheter-bedingte Komplikationen
Bei Patienten, die einen zentralen Venenkatheter benötigen, ist das Risiko für Katheter-bedingte Komplikationen zu berücksichtigen. Dazu gehören lokale Infektionen, Bakteriämie und Thrombosen im Bereich des Katheters.
Übertragbare Infektionserreger
Standardmaßnahmen zur Verhütung von Infektionen durch die Verabreichung von Arzneimitteln, die aus menschlichem Blut oder Plasma hergestellt wurden, sind Spenderauswahl, Testung einzelner Spenden und von Plasmapools auf spezifische Infektionsmarker und Einführung effektiver Herstellungsschritte zur Inaktivierung/Eliminierung von Viren. Dennoch kann bei Verabreichung von Arzneimitteln, die aus menschlichem Blut oder Plasma hergestellt wurden, die Möglichkeit der Übertragung von Erregern nicht völlig ausgeschlossen werden. Dies trifft auch für bisher unbekannte oder neu auftretende Viren und andere Erreger zu.
Die ergriffenen Maßnahmen werden als wirksam gegen umhüllte Viren wie Human-Immunodeficiency-Virus (HIV), Hepatitis-B-Virus (HBV) und Hepatitis-C-Virus (HCV) sowie gegen das nicht-umhüllte Hepatitis-A-Virus (HAV) angesehen. Die ergriffenen Maßnahmen sind möglicherweise bei nicht-umhüllten Viren wie Parvovirus B19 von begrenztem Wert.
Eine Parvovirus B19-Infektion kann schwere Erscheinungen bei schwangeren Frauen (fetale Infektion) und Patienten mit einer Immunschwäche oder verstärkter Erythropoese (z. B. bei hämolytischer Anämie) hervorrufen.
Bei Patienten, die regelmäßig/wiederholt aus Plasma vom Menschen gewonnene Faktor-VIII-Präparate erhalten, sollten entsprechende Impfungen (Hepatitis A und B) vorgesehen werden.
Es wird dringend empfohlen, bei jeder Behandlung mit Haemoctin SDH 250, 500 oder 1000 den Namen und die Chargenzeichnung des Präparats zu dokumentieren, um auf diese Weise eine Verbindung zwischen Patient und Produktcharge herzustellen.
Natriumgehalt
Eine Durchstechflasche enthält bis zu 1,4 mmol (32,2 mg) Natrium. Dies ist zu berücksichtigen bei Personen unter Natrium kontrollierter (natriumarmer/-kochsalzarmer) Diät.
Kinder und Jugendliche
Die für Erwachsene beschriebenen besonderen Warnhinweise und Vorsichtsmaßnahmen für die Anwendung sind auch bei Kindern und Jugendlichen zu beachten.
4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Es wurden keine Studien zur Erfassung von Wechselwirkungen durchgeführt.
Es wurden keine Wechselwirkungen zwischen Präparaten mit Blutgerinnungsfaktor VIII vom Menschen und anderen Arzneimitteln beschrieben.
4.6 Fertilität, Schwangerschaft und Stillzeit
Fertilität
Es liegen keine Daten zur Fertilität vor.
Schwangerschaft und Stillzeit
Reproduktionsstudien bei Tieren wurden mit Haemoctin SDH 250, 500 bzw. 1000 nicht durchgeführt. Aufgrund des seltenen Auftretens von Hämophilie A bei Frauen liegen über die Anwendung von Haemoctin SDH 250, 500 bzw. 1000 während der Schwangerschaft und Stillzeit keine Erfahrungen vor. Haemoctin SDH 250, 500 bzw. 1000 soll daher nur bei eindeutiger Indikationsstellung während Schwangerschaft und Stillzeit angewandt werden.
4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Haemoctin SDH 250, 500 oder 1000 hat keinen oder einen zu vernachlässigenden Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
4.8 Nebenwirkungen
Zusammenfassung des Sicherheitsprofils
Überempfindlichkeit oder allergische Reaktionen (wie z. B. Quincke-Ödem, Brennen und Stechen an der Injektionsstelle, Schüttelfrost, Hautrötungen, generalisierte Urtikaria, Kopfschmerz, Ausschlag, Hypotonie, Lethargie, Übelkeit, nervöse Unruhe, Tachykardie, Brustenge, Kribbeln, Erbrechen, Stridor) wurden selten beobachtet. Diese können in manchen Fällen zu schwerer Anaphylaxie (einschließlich Schock) führen.
Patienten mit Hämophilie A können neutralisierende Antikörper (Hemmkörper) gegen Faktor VIII entwickeln. Diese Situation manifestiert sich in unzureichender klinischer Wirksamkeit. Es wird empfohlen, in einem solchen Fall ein spezialisiertes Hämophilie-Zentrum zu kontaktieren.
Für Informationen über die Sicherheit im Hinblick auf übertragbare Erreger siehe Abschnitt 4.4.
Tabellarische Auflistung der Nebenwirkungen
Die unten dargestellte Tabelle entspricht der MedDRA-Systemorganklassen-Klassifikation (Systemorganklasse und Ebene der Bevorzugten Begriffe).
Bei der Bewertung von Nebenwirkungen werden folgende Häufigkeitsangaben zugrunde gelegt:
|
Sehr häufig: |
>1/10 |
|
Häufig: |
>1/100, <1/10 |
|
Gelegentlich: |
>1/1.000, <1/100 |
|
Selten: |
>1/10.000, <1/1.000 |
|
Sehr selten: |
<1/10.000, einschließlich Einzelfälle |
Im Zusammenhang mit Haemoctin SDH 250, 500 und 1000 wurden im Rahmen von klinischen Studien, Anwendungsbeobachtungen, Spontanmeldungen und routinemäßigen Literaturrecherchen die folgenden Nebenwirkungen berichtet:
|
Systemorganklassen gemäß MedDRA-Datenbank |
Nebenwirkungen |
Häufigkeit |
|
Erkrankungen des Nervensystems |
Hirnblutungen |
sehr selten |
|
Erkrankungen des Blutes und des Lymphsystems |
Anämie |
sehr selten |
|
Erkrankungen der Haut und des Unterhautzellgewebes |
Exanthem, Urtikaria, Erythem |
sehr selten |
Untersuchungen
Anti-Faktor VIII Antikörper positiv sehr selten
Beschreibung ausgewählter Nebenwirkungen
Überempfindlichkeit
Bei Patienten, die mit Faktor-VIII-haltigen Präparaten behandelt wurden, wurden Überempfindlichkeits- oder anaphylaktische Reaktionen beschrieben (mit beispielsweise Angioödem, Schüttelfrost, Flushing, Urtikaria, Kopfschmerzen, Hypotonie, Schmerzen an der Injektionsstelle, Lethargie, Übelkeit, Pruritus, Hautausschlag, Ruhelosigkeit, Tachykardie, Beschwerden im Brustkorb, Parästhesien, Fieber, Erbrechen, pfeifender Atem). In einigen Fällen entwickelten sich aus diesen Reaktionen schwere anaphylaktische Reaktionen (wie anaphylaktischer Schock).
Faktor-VIII-Hemmung
Patienten mit Hämophilie A können neutralisierende Antikörper (Hemmkörper) gegen Faktor-VIII-haltige Präparate entwickeln. Diese Situation manifestiert sich in unzureichender klinischer Wirksamkeit (z. B. Blutungen). Es wird empfohlen, in einem solchen Fall ein spezialisiertes Hämophilie-Zentrum zu kontaktieren. Das Auftreten von hemmenden Antikörpern ist eine bekannte Komplikation der Behandlung von Personen mit Hämophilie A.
Kinder und Jugendliche
Es wird erwartet, dass Häufigkeit, Art und Schweregrad von Nebenwirkungen bei Kindern und Jugendlichen denen bei Erwachsenen entsprechen.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel, Paul-Ehrlich-Institut, Paul-Ehrlich-Str. 51-59, 63225 Langen, Tel: +49 6103 77 0, Fax: +49 6103 77 1234, Website: www.pei.de, anzuzeigen.
4.9 Überdosierung
Es wurden keine Fälle von Überdosierung berichtet.
5. PHARMAKOLOGISCHE EIGENSCHAFTEN
5.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Antihämorrhagika: Blutgerinnungsfaktor VIII ATC-Code: B02BD02.
Der Faktor VIII/von-Willebrand-Faktor(vWF)-Komplex besteht aus zwei Molekülen (Faktor VIII und von-Willebrand-Faktor) mit unterschiedlichen physiologischen Funktionen. Wird einem Hämophilie-Patienten Faktor VIII injiziert, so bindet dieser im Blutkreislauf an den von-Willebrand-Faktor.
Der aktivierte Faktor VIII wirkt als Cofaktor für den aktivierten Faktor IX und beschleunigt die Bildung von aktiviertem Faktor X (Faktor Xa) aus Faktor X. Faktor Xa aktiviert Prothrombin zu Thrombin. Dieses setzt dann aus Fibrinogen Fibrin frei und die Gerinnselbildung kann erfolgen. Hämophilie A ist eine geschlechtsgebundene erbliche Störung der Blutgerinnung aufgrund erniedrigter Faktor-VIII:C-Plasmaspiegel. Dies führt entweder spontan oder in Folge unfallbedingter oder chirurgischer Traumata zu starken Blutungen in Gelenken, Muskeln oder inneren Organen. Durch die Substitutionstherapie werden die Faktor-VIII-Plasmaspiegel erhöht, wodurch eine temporäre Korrektur des Faktor-VIII-Mangels ermöglicht und die Blutungstendenz korrigiert wird.
Zusätzlich zur Rolle des Faktor-VIII-schützenden Proteins vermittelt der von-Willebrand-Faktor das Haften der Thrombozyten bei Gefäßverletzungen und spielt eine Rolle bei der Thrombozytenaggregation.
5.2 Pharmakokinetische Eigenschaften
Nach intravenöser Gabe nimmt die Faktor-VIII-Aktivität exponentiell biphasisch ab. In der Initialphase vollzieht sich die Verteilung zwischen dem intravaskulären Raum und den übrigen Verteilungsräumen (Körperflüssigkeiten) mit einer Plasma-Halbwertszeit von 1 bis 8 Stunden In der nachfolgenden Phase liegt die Halbwertszeit zwischen 5 und 18 Stunden, mit einem Mittel von ca. 12 Stunden. Dies entspricht offenbar der physiologischen Halbwertszeit.
Die „incremental recovery“ von Haemoctin SDH 250, 500 bzw. 1000 beträgt ca. 0,020 ± 0,003 I.E./ml/I.E./kg KG. Nach intravenöser Gabe von 1 I.E. Faktor VIII pro kg KG beträgt die Faktor-VIII-Aktivität etwa 2 %.
Weitere pharmakokinetische Parameter für Haemoctin SDH 250, 500 bzw. 1000 sind:
• Area under the curve (AUC): ca. 17 I.E. * h/ml
• Mittlere Verweildauer (MRT): ca. 15 h
• Clearance: ca. 155 ml/h
5.3 Präklinische Daten zur Sicherheit
Blutgerinnungsfaktor VIII vom Menschen (aus dem Konzentrat) ist ein normaler Bestandteil des menschlichen Plasmas und unterscheidet sich in seinen Wirkungen nicht von dem endogenen Faktor VIII.
Prüfungen der Toxizität bei einmaliger Verabreichung sind nicht aussagekräftig, da höhere Dosen bei den Tieren zu einer Volumenüberlastung führen. Toxizitätsprüfungen mit wiederholter Verabreichung sind im Tierversuch aufgrund der Antikörperbildung gegen heterologes Protein nicht durchführbar.
Auch das Mehrfache der für den Menschen pro Kilogramm Körpergewicht empfohlenen Dosis zeigt bei Labortieren keine toxischen Wirkungen.
Da es im Zusammenhang mit der klinischen Anwendung von Blutgerinnungsfaktor VIII vom Menschen bisher keine Hinweise auf kanzerogene und mutagene Wirkungen gibt, werden entsprechende Tierversuche nicht als erforderlich angesehen.
6. PHARMAZEUTISCHE ANGABEN
6.1 Liste der sonstigen Bestandteile
Pulver: Glycin, Natriumchlorid, Natriumcitrat, Calciumchlorid Lösungsmittel: Wasser für Injektionszwecke
6.2 Inkompatibilitäten
Da keine Kompatibilitätsstudien durchgeführt wurden, darf Haemoctin SDH 250, 500 bzw.
1000 nicht mit anderen Arzneimitteln gemischt werden.
Es sollte nur das beigefügte Infusionsbesteck verwendet werden, da ein Therapieversagen aufgrund der Adsorption von Faktor VIII an den Innenflächen einiger anderer Infusionsbestecke auftreten kann.
6.3 Dauer der Haltbarkeit
2 Jahre
Das Präparat soll nach dem ersten Öffnen sofort verabreicht werden.
6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Nicht über 25°C lagern. Nicht einfrieren.
Durchstechflaschen im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
6.5 Art und Inhalt des Behältnisses
1 Packungseinheit. Haemoctin SDH 250, 500 bzw. 1000 enthält:
1 Durchstechflasche mit Pulver (20 ml) Glastyp I gemäß Ph.Eur.
Gefriertrocknungs-Stopfen aus Halobutyl-Kautschuk Typ I gemäß Ph.Eur..
1 Durchstechflasche mit Lösungsmittel (5 ml, 10 ml) Glastyp I gemäß Ph.Eur.. InjektionsStopfen aus Halobutyl-Kautschuk Typ I gemäß Ph.Eur..
Die Packung enthält zusätzlich:
1 Einmalspritze (5 ml, 10 ml), 1 Transfersystem mit integriertem Filter, 1 Flügelkanüle und 2 sterile Alkoholtupfer.
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung
Das rekonstituierte Arzneimittel muss vor der Verabreichung visuell auf Partikel und Verfärbung überprüft werden. Die Lösung muss klar oder leicht opaleszierend sein. Verabreichen Sie keine Lösungen, die trüb (wolkig) sind oder Ablagerungen enthalten.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Hinweise für die Anwendung und Handhabung:
Bei sämtlichen Arbeitsvorgängen ist auf aseptische Bedingungen zu achten!
Fig. 1
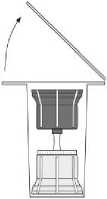
Fig. 2
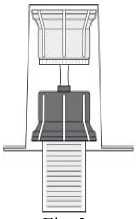
Fig. 3
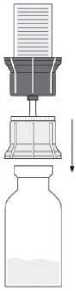
Fig. 4

Fig. 5
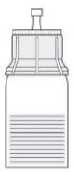
Fig. 6

Fig. 7
Erwärmen Sie Lösungsmittel (Wasser für Injektionszwecke) und Pulver in den ungeöffneten Durchstechflaschen auf Zimmertemperatur. Wird zum Erwärmen ein Wasserbad benutzt, muss sorgfältig darauf geachtet werden, dass das Wasser nicht mit den Kappen oder Stopfen der Durchstechflaschen in Berührung kommt. Andernfalls kann es zur Kontamination des Arzneimittels kommen.
Entfernen Sie die Kappen von beiden Durchstechflaschen, um den mittleren Teil des Gummistopfens freizulegen (1). Reinigen Sie die Gummistopfen der Durchstechflaschen für das Pulver und das Lösungsmittel mit einem Desinfektionsmittel.
Entfernen Sie die Oberseite der Verpackung des Transfersystems (2). Setzen Sie den blauen Teil des Transfersystems auf die aufrecht stehende Durchstechflasche mit dem Lösungsmittel (3).
Nehmen Sie das Transfersystem ganz aus der Verpackung. Jetzt erscheint der transparente Teil des Transfersystems.
Stellen Sie die Durchstechflasche mit dem Pulver auf eine ebene Fläche.
Drehen Sie die Einheit aus dem Transfersystem und der Durchstechflasche mit dem Lösungsmittel auf den Kopf und stechen Sie den Adapter mit dem Dorn seines transparenten Teils senkrecht in den Stopfen der aufrecht stehenden Durchstechflasche mit dem Pulver (4). Durch das in der Durchstechflasche mit dem Pulver vorhandene Vakuum läuft das Wasser in diese Durchstechflasche (5). Drehen Sie sofort den blauen Teil des Transfersystems zusammen mit der Durchstechflasche mit dem Lösungsmittel ab und entsorgen Sie diese, ohne sie zu trennen (6). Vorsichtiges Schwenken des Präparats hilft beim Auflösen des Pulvers. Bitte nicht kräftig schütteln, jegliche Schaumbildung ist zu vermeiden! Die Lösung ist klar oder leicht opaleszierend (milchig glänzend).
Die gebrauchsfertige Lösung soll unmittelbar nach der Auflösung verwendet werden. Verwenden Sie keine Lösungen, die trüb (wolkig) sind oder sichtbare Partikel enthalten.
Injektion:
Nach Lösung des Pulvers (wie oben beschrieben) die beigefügte Spritze mit dem Luer-Lock-Anschluss auf den transparenten Teil des Transfersystems schrauben, der noch in der Durchstechflasche mit dem gelösten Pulver steckt (7). Anschließend lässt sich das gelöste Präparat problemlos in die Spritze aufziehen. Ein separater Filter ist nicht nötig, da das Transfersystem einen integrierten Filter besitzt.
Die Durchstechflasche mit dem transparenten Teil des Transfersystems vorsichtig von der Spritze abschrauben. Die Injektionslösung mit der beigefügten Flügelkanüle sofort langsam intravenös injizieren. Die Injektionsgeschwindigkeit darf 2 - 3 ml pro Minute nicht überschreiten.
Nach Gebrauch der Flügelkanüle kann deren Nadel durch die Schutzkappe gesichert werden.
7. INHABER DER ZULASSUNG
Biotest Pharma GmbH Landsteinerstraße 5 63303 Dreieich Deutschland Tel.: 06103 801-0 Fax: 06103 801-150
8. ZULASSUNGSNUMMER(N)
Haemoctin SDH 250: 16841.00.00
Haemoctin SDH 500: 16841.01.00
Haemoctin® SDH 1000: 16841.02.00
9. DATUM DER ERTEILUNG DER ZULASSUNG/VERLÄNGERUNG DER ZULASSUNG
07.11.1991/18.03.2013
10. STAND DER INFORMATION
12/2014
11. VERSCHREIBUNGSSTATUS / APOTHEKENPFLICHT
Verschreibungspflichtig
12. SONSTGE HINWEISE
Herkunftsländer des Blutplasmas:
Belgien, Deutschland, Niederlande, Österreich, Schweiz, Tschechische Republik, Ungarn, USA Es wird auf die Dokumentationspflicht gemäß Transfusionsgesetz hingewiesen.
10 / 10