Haemoctin Sdh 500
Gebrauchsinformation: Information für Anwender
Haemoctin SDH 500
Pulver und Lösungsmittel zur Herstellung einer Injektionslösung Aus Plasma vom Menschen gewonnener Blutgerinnungsfaktor VIII
Lesen Sie die gesamte Packungsbeilage sorgfältig durch, bevor Sie mit der Anwendung dieses Arzneimittels beginnen, denn sie enthält wichtige Informationen.
- Heben Sie die Packungsbeilage auf. Vielleicht möchten Sie diese später nochmals lesen.
- Wenn Sie weitere Fragen haben, wenden Sie sich an Ihren Arzt, Apotheker oder das medizinische Fachpersonal.
- Dieses Arzneimittel wurde Ihnen persönlich verschrieben. Geben Sie es nicht an Dritte weiter. Es kann anderen Menschen schaden, auch wenn diese die gleichen Beschwerden haben wie Sie.
- Wenn Sie Nebenwirkungen bemerken, wenden Sie sich an Ihren Arzt, Apotheker oder das medizinische Fachpersonal. Dies gilt auch für Nebenwirkungen, die nicht in dieser Packungsbeilage angegeben sind. Siehe Abschnitt 4.
Was in dieser Packungsbeilage steht:
1. Was ist Haemoctin SDH 500 und wofür wird es angewendet?
2. Was sollten Sie vor der Anwendung von Haemoctin SDH 500 beachten?
3. Wie ist Haemoctin SDH 500 anzuwenden?
4. Welche Nebenwirkungen sind möglich?
5. Wie ist Haemoctin SDH 500 aufzubewahren?
6. Inhalt der Packung und weitere Informationen
1. Was ist Haemoctin SDH 500 und wofür wird es angewendet?
Haemoctin SDH 500 ist ein aus menschlichem Blutplasma hergestelltes Arzneimittel. Es enthält den Blutgerinnungsfaktor VIII, der für den normalen Ablauf der Blutgerinnung erforderlich ist. Nach dem Auflösen des Pulvers mit 10 ml Wasser für Injektionszwecke ist die Lösung zur intravenösen Injektion bereit.
Haemoctin SDH 500 wird angewendet zur Behandlung und Vorbeugung von Blutungen bei angeborenem Mangel an Gerinnungsfaktor VIII (Hämophilie A).
Haemoctin SDH 500 enthält den von-Willebrand-Faktor nicht in pharmakologisch wirksamer Menge und ist daher nicht zur Behandlung der von-Willebrand-Erkrankung geeignet.
2. Was sollten Sie vor der Anwendung von Haemoctin SDH 500 beachten?
Haemoctin SDH 500 darf nicht angewendet werden,
- wenn Sie allergisch (überempfindlich) gegen Blutgerinnungsfaktor VIII oder einen der in Abschnitt 6. genannten sonstigen Bestandteile dieses Arzneimittels sind. Eine allergische Reaktion kann Ausschlag, Jucken, Atemschwierigkeiten, Schwellung des Gesichts, der Lippen, des Rachens oder der Zunge beinhalten.
Warnhinweise und Vorsichtsmaßnahmen
Bitte sprechen Sie mit Ihrem Arzt, bevor Sie Haemoctin SDH 500 anwenden.
Wenn Sie die Behandlung mit Haemoctin SDH 500 beginnen, ist es möglich, dass Ihr Immunsystem Antikörper (Hemmkörper) gegen Faktor VIII entwickelt.
Diese Hemmkörper können die Wirkung von Haemoctin SDH 500 beeinträchtigen. Ihr Arzt sollte regelmäßig mit einem biologischen Test (Bethesda-Test) untersuchen, ob bei Ihnen eine Bildung von Hemmkörpern stattgefunden hat. Das Auftreten solcher Faktor-VIII-Hemmkörper zeigt sich durch Ausbleiben des Behandlungserfolgs. Die Hemmkörpermenge wird in Bethesda-Einheiten (BE) pro ml Blutplasma angegeben. Das Risiko, Hemmkörper zu entwickeln, ist abhängig von der Häufigkeit der Gabe von Faktor VIII, wobei dieses Risiko während der ersten 20 Anwendungstage am größten ist.
Selten bilden sich Hemmkörper nach mehr als 100 Anwendungstagen. Fälle wiederkehrender Hemmkörper wurden nach Wechsel von einem Faktor-VIII-Präparat auf ein anderes bei vorbehandelten Patienten mit mehr als 100 Expositionstagen, die zuvor Hemmkörper gebildet hatten, beobachtet.
Es sollte nur das beigefügte Infusionsbesteck verwendet werden, da ein Ausbleiben des Behandlungserfolgs aufgrund der Anlagerung (Adsorption) von Faktor VIII an den Innenflächen bestimmter Infusionsbestecke auftreten kann.
Haemoctin SDH 500 darf nicht mit anderen Arzneimitteln gemischt werden.
Katheter-bedingte Komplikationen: Wenn ein zentraler Venenkatheter benötigt wird, ist das Risiko für Katheter-bedingte Komplikationen zu berücksichtigen. Dazu gehören lokale Infektionen, Bakterien im Blut (Bakteriämie) und Thrombosen im Bereich des Katheters.
Virussicherheit
Bei der Herstellung von Arzneimitteln aus menschlichem Blut oder Blutplasma werden bestimmte Maßnahmen ergriffen, um zu vermeiden, dass Infektionserreger auf Patienten übertragen werden. Zu diesen Maßnahmen zählt:
- die sorgfältige Auswahl von Blut- und Plasmaspendern. Auf diese Weise möchte man sicherstellen, dass Personen, die möglicherweise mit Krankheitserregern infiziert sind, ausgeschlossen werden.
- das Prüfen jeder Spende und jedes Plasmapools auf Anzeichen von Viren/Infektionen.
- die Einführung von Arbeitsschritten in die Verarbeitung von Blut oder Plasma, die zur Inaktivierung oder Entfernung von Viren führen.
Trotz dieser Maßnahmen kann bei der Verabreichung von Arzneimitteln, die aus menschlichem Blut oder Plasma hergestellt werden, das Risiko einer Übertragung von Infektionen nicht vollständig ausgeschlossen werden. Dies gilt auch für unbekannte oder neu auftretende Viren und andere Infektionserreger.
Die ergriffenen Maßnahmen werden als wirksam gegenüber umhüllten Viren erachtet, wie das menschliche Immunschwächevirus (HIV), das Hepatitis-B-Virus und das Hepatitis-C-Virus, und gegen das nicht umhüllte Hepatitis-A-Virus. Die ergriffenen Maßnahmen sind möglicherweise bei anderen nicht umhüllten Viren wie dem Parvovirus B19 nur von begrenztem Wert. Eine Parvovirus-B19-Infektion kann schwere Erscheinungen bei schwangeren Frauen (Infektion des ungeborenen Kindes) und Patienten mit einer Immunschwäche oder einigen Arten von Blutarmut(z. B. Sichelzellen-Krankheit oder hämolytische Anämie) hervorrufen.
Ihr Arzt wird Ihnen empfehlen, eine Impfung gegen Hepatitis A und B in Betracht zu ziehen, wenn Sie regelmäßig/wiederholt Faktor-VIII-Präparate aus menschlichem Blutplasma erhalten.
Es wird dringend empfohlen, bei jeder Behandlung mit Haemoctin SDH 500 den Namen und die Chargenbezeichnung des Arzneimittels zu vermerken, um die Rückverfolgbarkeit sicherzustellen.
Anwendung von Haemoctin SDH 500 zusammen mit anderen Arzneimitteln
Informieren Sie Ihren Arzt, wenn Sie andere Arzneimittel anwenden, kürzlich andere Arzneimittel angewendet haben oder beabsichtigen, andere Arzneimittel anzuwenden.
Wechselwirkungen zwischen Haemoctin SDH 500 und anderen Arzneimitteln sind nicht bekannt. Schwangerschaft und Stillzeit
Wenn Sie schwanger sind oder stillen, oder wenn Sie vermuten, schwanger zu sein oder beabsichtigen, schwanger zu werden, fragen Sie vor der Anwendung dieses Arzneimittels Ihren Arzt um Rat. Aufgrund des seltenen Auftretens der Hämophilie A bei Frauen liegen über die Anwendung von Haemoctin SDH 500 während Schwangerschaft und Stillzeit keine Erfahrungen vor. Es wurden keine tierexperimentellen Untersuchungen in der Schwangerschaft und während der Stillzeit durchgeführt.
Verkehrstüchtigkeit und das Bedienen von Maschinen
Die Fähigkeit zur aktiven Teilnahme am Straßenverkehr und zum Bedienen von Maschinen wird durch die Anwendung von Haemoctin SDH 500 nicht beeinträchtigt.
Haemoctin SDH 500 enthält Natrium
Eine Durchstechflasche enthält bis zu 1,40 mmol (32,2 mg) Natrium. Wenn Sie eine kochsalzarme Diät einhalten müssen, sollten Sie dies berücksichtigen.
3. Wie ist Haemoctin SDH 500 anzuwenden?
Haemoctin SDH 500 ist zur intravenösen Anwendung (Injektion in eine Vene) bestimmt. Die Behandlung muss unter Überwachung eines Arztes begonnen werden, der mit der Therapie der Hämophilie A vertraut ist. Wenden Sie Haemoctin SDH 500 immer genau nach Absprache mit Ihrem Arzt an. Fragen Sie bei Ihrem Arzt nach, wenn Sie sich nicht sicher sind.
Dosierung und Dauer der Behandlung sind abhängig von der Schwere des Faktor-VIII-Mangels. Zudem sind der Ort und das Ausmaß der Blutung entscheidend, außerdem Ihr allgemeiner Gesundheitszustand. Ihr Arzt wird die für Sie geeignete Dosis bestimmen.
Bei sämtlichen Arbeitsvorgängen ist auf sterile Bedingungen zu achten.
Fig. 1
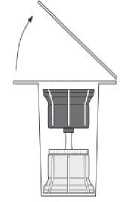
Fig. 2
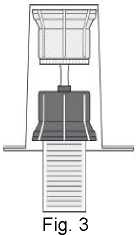

Fig. 5
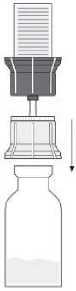
Fig. 4
Fig. 6
Lösen des Pulvers:
- Erwärmen Sie Lösungsmittel (Wasser für Injektionszwecke) und Pulver in den ungeöffneten Durchstechflaschen auf Zimmertemperatur. Wird zum Erwärmen ein Wasserbad benutzt, muss sorgfältig darauf geachtet werden, dass das Wasser nicht mit den Kappen oder Stopfen der Durchstechflaschen in Berührung kommt. Andernfalls kann es zu Verunreinigungen des Arzneimittels kommen.
- Entfernen Sie die Kappen von beiden Durchstechflachen, um den mittleren Teil des Gummistopfens freizulegen (1). Reinigen Sie die Gummistopfen der Durchstechflaschen für das Pulver und das Lösungsmittel mit einem Desinfektionsmittel.
- Entfernen Sie die Oberseite der Verpackung des Transfersystems (2). Setzen Sie den blauen Teil des Transfersystems auf die aufrecht stehende Durchstechflasche mit dem Lösungsmittel (3).
- Nehmen Sie das Transfersystem ganz aus der Verpackung. Jetzt erscheint der transparente Teil des Transfersystems.
- Stellen Sie die Durchstechflasche mit dem Pulver auf eine ebene Fläche.
- Drehen Sie die Einheit aus dem Transfersystem und der Durchstechflasche mit dem Lösungsmittel auf den Kopf und stechen Sie den Adapter mit dem Dorn seines transparenten Teils senkrecht in den Stopfen der aufrecht stehenden Durchstechflasche mit dem Pulver (4). Durch das in der Durchstechflasche mit dem Pulver vorhandene Vakuum läuft das Wasser in diese Durchstechflasche (5). Drehen Sie sofort den blauen Teil des Transfersystems zusammen mit der Durchstechflasche mit dem Lösungsmittel ab und entsorgen Sie diese, ohne sie zu trennen (6). Vorsichtiges Schwenken des Präparats hilft beim Auflösen des Pulvers. Bitte nicht kräftig schütteln, jegliche Schaumbildung ist zu vermeiden! Die Lösung ist klar oder leicht opaleszierend (milchig glänzend).
- Die gebrauchsfertige Lösung soll unmittelbar nach der Auflösung verwendet werden. Verwenden Sie keine Lösungen, die trüb (wolkig) sind oder sichtbare Partikel enthalten.

Injektion:
- Nach Lösung des Pulvers (wie oben beschrieben) die beigefügte Spritze mit dem Luer-Lock-Anschluss auf den transparenten Teil des Transfersystems schrauben, der noch in der Durchstechflasche mit dem gelösten Pulver steckt. (7) Anschließend lässt sich das gelöste Präparat problemlos in die Spritze aufziehen. Ein separater Filter ist nicht nötig, da das Transfersystem einen integrierten Filter besitzt.
- Die Durchstechflasche mit dem transparenten Teil des Transfersystems vorsichtig von der Spritze abschrauben. Die Injektionslösung mit der beigefügten Flügelkanüle sofort langsam intravenös injizieren. Es wird empfohlen, nicht mehr als 2 - 3 ml/min zu verabreichen.
- Nach Gebrauch der Flügelkanüle kann deren Nadel durch die Schutzkappe gesichert werden.
Wenn Sie weitere Fragen zur Anwendung dieses Arzneimittels haben, wenden Sie sich an Ihren Arzt oder Apotheker.
Wenn Sie eine größere Menge von Haemoctin SDH 500 angewendet haben, als Sie sollten
Wenn Sie meinen, dass Ihnen zu viel Haemoctin SDH 500 gegeben wurde, informieren Sie Ihren Arzt,
der über die weitere Behandlung entscheiden wird.
Wenn Sie die Anwendung von Haemoctin SDH 500 vergessen haben
In diesem Fall wird Ihr Arzt entscheiden, ob eine weitere Behandlung erforderlich ist.
Wenn Sie die Anwendung von Haemoctin SDH 500 abbrechen
Brechen Sie die Anwendung von Haemoctin SDH 500 nicht ohne Rücksprache mit Ihrem Arzt ab.
4. Welche Nebenwirkungen sind möglich?
Wie alle Arzneimittel kann auch dieses Arzneimittel Nebenwirkungen haben, die aber nicht bei jedem auftreten müssen.
Informieren Sie umgehend Ihren Arzt, wenn Sie eine der folgenden Nebenwirkungen bemerken:
- gerötete Haut
- Brennen und Stechen an der Injektionsstelle
- Schüttelfrost
- plötzliche Hautrötungen
- Kopfschmerzen
- Nesselsucht
- niedriger Blutdruck
- Trägheit
- Übelkeit
- Unruhe
- schneller Herzschlag
- Engegefühl in der Brust
- Kribbeln
- Erbrechen
- pfeifender Atem
Hierbei kann es sich um eine allergische oder schwere allergische Reaktion (anaphylaktischer Schock) oder eine Überempflichkeitsreaktion handeln.
Die folgenden anderen Nebenwirkungen wurden sehr selten (betrifft weniger als 1 Anwender von 10.000) berichtet:
- Anti-Faktor VIII Antikörper positiv: Patienten mit Hämophilie A können neutralisierende Antikörper (Hemmkörper) gegen Faktor-VIII entwickeln. Das Auftreten solcher Hemmkörper zeigt sich durch Ausbleiben des Behandlungserfolgs (z. B. Blutungen). Es wird empfohlen, in einem solchen Fall ein spezialisiertes Hämophilie-Zentrum zu kontaktieren.
- Fieber
- Blutarmut (Anämie)
- Hirnblutungen
Nebenwirkungen bei Kindern und Jugendlichen
Es wird erwartet, dass die Nebenwirkungen bei Kindern denen bei Erwachsenen entsprechen. Meldung von Nebenwirkungen
Wenn Sie Nebenwirkungen bemerken, wenden Sie sich an Ihren Arzt, Apotheker oder das medizinische Fachpersonal. Dies gilt auch für Nebenwirkungen, die nicht in dieser Packungsbeilage angegeben sind. Sie können Nebenwirkungen auch direkt über das nationale Meldesystem anzeigen:
Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel
Paul-Ehrlich-Institut
Paul-Ehrlich-Str. 51-59
63225 Langen
Tel: +49 6103 77 0
Fax: +49 6103 77 1234
Website: www.pei.de
Indem Sie Nebenwirkungen melden, können Sie dazu beitragen, dass mehr Informationen über die Sicherheit dieses Arzneimittels zur Verfügung gestellt werden.
5. Wie ist Haemoctin SDH 500 aufzubewahren?
Bewahren Sie dieses Arzneimittel für Kinder unzugänglich auf.
Durchstechflaschen im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Nicht über 25°C lagern. Nicht einfrieren.
Sie dürfen dieses Arzneimittel nach dem auf dem Etikett der Durchstechflasche und dem Umkarton angegebenen Verfalldatum nicht mehr verwenden.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu entsorgen. Entsorgen Sie Arzneimittel nicht im Abwasser oder Haushaltsabfall. Fragen Sie Ihren Apotheker, wie das Arzneimittel zu entsorgen ist, wenn Sie es nicht mehr verwenden. Sie tragen damit zum Schutz der Umwelt bei.
6. Inhalt der Packung und weitere Informationen Was Haemoctin SDH 500 enthält
- Der Wirkstoff ist: Blutgerinnungsfaktor VIII vom Menschen
- Die sonstigen Bestandteile sind: Glycin, Natriumchlorid, Natriumcitrat und Calciumchlorid
- Die Durchstechflasche mit dem Lösungsmittel enthält Wasser für Injektionszwecke.
Wie Haemoctin SDH 500 aussieht und Inhalt der Packung
Haemoctin SDH 500 liegt als gefriergetrocknetes Pulver (Lyophilisat) vor. Wasser für Injektionszwecke dient als Lösungsmittel. Das aufgelöste Arzneimittel ist klar oder leicht opaleszierend (milchig glänzend).
Weitere Packungsgrößen:
Haemoctin SDH 250: enthält 1 Durchstechflasche mit 250 I.E. und 1 Durchstechflasche mit 5 ml Wasser für Injektionszwecke (50 I.E./ml)
Haemoctin SDH 1000: enthält 1 Durchstechflasche mit 1000 I.E. und 1 Durchstechflasche mit 10 ml Wasser für Injektionszwecke (100 I.E./ml)
Jede Packung enthält:
- eine Einmalspritze
- ein Transfersystem mit integriertem Filter
- eine Flügelkanüle
- zwei sterile Alkoholtupfer
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
Pharmazeutischer Unternehmer und Hersteller
Biotest Pharma GmbH Landsteinerstraße 5 63303 Dreieich Deutschland Tel.: 06103 801-0 Fax: 06103 801-150 mail@biotest.de
Dieses Arzneimittel ist in den Mitgliedstaaten des Europäischen Wirtschaftsraumes (EWR) unter den folgenden Bezeichnungen zugelassen:
Deutschland, Litauen, Malta, Rumänien, Tschechische Republik: Haemoctin® SDH 500
Belgien, Griechenland, Italien, Niederlande, Polen, Portugal, Spanien, Vereinigtes Königreich: Haemoctin® 500
Bulgarien: Haemoctin® 500 IU
Österreich: Haemoctin® SDH 50 I.E./ml
Ungarn: Haemoctin® 500 NE
Diese Gebrauchsinformation wurde zuletzt überarbeitet 12/2014.
Herkunftsländer des Plasmas
Zur Herstellung von Haemoctin SDH 500 wird Blutplasma aus Belgien, Deutschland, Niederlande, Österreich, Schweiz, Tschechische Republik, Ungarn und USA verwendet.
Die folgenden Informationen sind für medizinisches Fachpersonal bestimmt:
Art der Anwendung
Haemoctin SDH 500 ist zur intravenösen Injektion bestimmt. Es wird empfohlen, nicht mehr als 2-3 ml pro Minute zu verabreichen.
Dosierung
Dosierung und Dauer der Substitutionstherapie sind abhängig von der Schwere des Faktor-VIII-Mangels sowie von Lokalisation und Ausmaß der Blutung und vom klinischen Zustand des Patienten. Die verabreichten Faktor-VIII-Einheiten werden in Internationalen Einheiten (I.E.) angegeben, abgeleitet vom aktuellen WHO-Standard für Faktor-VIII-Produkte. Die Faktor-VIII-Aktivität im Plasma wird entweder als Prozentsatz (bezogen auf normales Humanplasma) oder in Internationalen Einheiten (bezogen auf einen Internationalen Standard für Faktor VIII im Plasma) angegeben. Eine Internationale Einheit (I.E.) der Faktor-VIII-Aktivität entspricht der Menge an Faktor VIII in einem ml normalem menschlichen Plasma.
Die Berechnung der erforderlichen Faktor-VIII-Dosierung basiert auf dem empirischen Befund, dass die Gabe von 1 Internationaler Einheit (I.E.) Faktor VIII pro kg Körpergewicht die Faktor-VIII-Aktivität im Plasma um 1%-2%, bezogen auf den Normalwert, anhebt.
Die erforderliche Dosis wird mit der folgenden Formel berechnet:
Benötigte Einheiten = Körpergewicht (kg) x gewünschter Faktor-VTO-Anstieg (%) x 0,5
Dosis und Häufigkeit der Verabreichung sollten sich immer an der klinischen Wirksamkeit im jeweiligen Einzelfall orientieren.
Im Fall der aufgeführten Blutungsereignisse sollte die Faktor-VIII-Aktivität im entsprechenden Zeitraum nicht unter das angegebene Aktivitäts-Niveau im Plasma (in % der Norm) fallen. Die folgende Tabelle kann als Richtlinie für die Dosierung bei Blutungsereignissen und chirurgischen Eingriffen dienen:
|
Schwere der Blutung / Art des chirurgischen Eingriffs |
Benötigter Faktor-VIII-Plasmaspiegel (%) |
Häufigkeit der Dosierung (Stunden) / Behandlungsdauer (Tage) |
|
Blutungen | ||
|
Gelenkblutungen im Frühstadium, Muskelblutungen, Blutungen im Mundbereich |
20-40 |
Injektion alle 12-24 Stunden/ mindestens 1 Tag, bis die (durch Schmerzen erkennbare) Blutung sistiert bzw. Wundheilung erreicht ist. |
|
Ausgeprägtere Gelenkblutungen, Muskelblutungen oder Hämatome |
30-60 |
Injektion alle 12-24 Stunden für 3-4 Tage oder länger, bis Schmerzen und akute Behinderungen beseitigt sind. |
|
Lebensbedrohliche Blutungen |
60-100 |
Injektion alle 8-24 Stunden, bis die Gefahr vorüber ist. |
|
Chirurgische Eingriffe | ||
|
Kleinere Eingriffe einschließlich Zahnextraktionen |
30-60 |
Injektion alle 24 Stunden/mindestens 1 Tag, bis Wundheilung erreicht ist. |
|
Größere Eingriffe |
80-100 (prä- und postoperativ) |
Injektion alle 8-24 Stunden, bis ausreichende Wundheilung erreicht ist; dann für mindestens weitere 7 Tage einen Faktor-VIII-Spiegel von 30-60% aufrechterhalten. |
Während des Behandlungsverlaufs wird, zur Steuerung der zu verabreichenden Dosis und der Häufigkeit der Injektionen, eine angemessene Bestimmung der Faktor-VIII-Plasmaspiegel angeraten. Besonders bei größeren chirurgischen Eingriffen ist eine genaue Überwachung der Substitutionstherapie durch Bestimmung des Blutgerinnungsstatus (Faktor-VIII-Aktivität) unerlässlich. Einzelne Patienten können sich in ihrer Reaktion auf Faktor VIII unterscheiden, verschiedene /n-vivo-Wiederfindungsraten erreichen und unterschiedliche Halbwertszeiten aufweisen. Bei der Langzeitprophylaxe von Blutungen bei Patienten mit schwerer Hämophilie A beträgt die übliche Dosis 20 - 40 I.E. Faktor VIII pro kg Körpergewicht im Abstand von 2 - 3 Tagen. In manchen Fällen, insbesondere bei jüngeren Patienten, können kürzere Dosierungsintervalle oder höhere Dosen erforderlich sein.
Patienten sollten auf die Bildung von Hemmkörpern gegen Faktor VIII überwacht werden. Falls die erwarteten Faktor-VIII-Aktivitäten nicht erreicht werden oder die Blutung mit einer angemessenen Dosis nicht zu beherrschen ist, muss ein Hemmkörpertest durchgeführt werden. Bei Patienten mit hohen Hemmkörperspiegeln ist die Faktor-VIII-Therapie möglicherweise nicht wirksam und es sollten therapeutische Alternativen in Erwägung gezogen werden. Diese Patienten sollten nur unter Aufsicht eines Arztes behandelt werden, der Erfahrung mit der Behandlung von Hämophilie-Patienten hat.
Es wird auf die Dokumentationspflicht gemäß Transfusionsgesetz hingewiesen.
8/8