Iluvien 190 Mikrogramm Intravitreales Implantat Im Applikator
ZUSAMMENFASSUNG DER MERKMALE DES ARZNEIMITTELS
1. BEZEICHNUNG DES ARZNEIMITTELS
ILUVIEN 190 Mikrogramm intravitreales Implantat im Applikator
2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
Jedes Implantat enthält 190 Mikrogramm Fluocinolonacetonid.
Vollständige Auflistung der sonstigen Bestandteile siehe Abschnitt 6.1.
3. DARREICHUNGSFORM
Intravitreales Implantat im Applikator.
Hellbraunes, zylindrisches Implantat mit einer Größe von etwa 3,5 mm x 0,37 mm.
Implantat-Applikator mit einer Injektionsnadel 25 Gauge (G).
4. KLINISCHE ANGABEN
4.1 Anwendungsgebiete
ILUVIEN ist zur Behandlung von Sehstörungen in Verbindung mit chronischem diabetischem Makulaödem indiziert, das auf verfügbare Therapien nur unzureichend anspricht.
4.2 Dosierung, Art und Dauer der Anwendung
Dosierung
Die empfohlene Dosis ist ein ILUVIEN Implantat in das betroffene Auge. Die gleichzeitige Anwendung an beiden Augen wird nicht empfohlen (siehe Abschnitt 4.4).
Nach 12 Monaten kann ein weiteres Implantat eingesetzt werden, wenn bei dem Patienten eine Sehverschlechterung auftritt oder in Verbindung mit einem rezidivierenden oder verschlimmerten diabetischen Makulaödem die Netzhautdicke zunimmt (siehe Abschnitt 5.1).
Eine erneute Behandlung sollte nur dann in Betracht gezogen werden, wenn der potenzielle Nutzen das Risiko rechtfertigt.
Mit ILUVIEN sollten nur Patienten behandelt werden, die auf eine vorherige Laserphotokoagulationstherapie oder andere verfügbare Therapien für diabetisches Makulaödem nur unzureichend angesprochen haben.
Kinder und Jugendliche
Es gibt im Anwendungsgebiet diabetisches Makulaödem (DMÖ) keinen relevanten Nutzen von ILUVIEN bei Kindern und Jugendlichen.
Spezielle Populationen
Bei älteren Patienten und bei Patienten mit Nieren- oder Leberfunktionsstörungen ist keine Dosierungsanpassung erforderlich.
Art der Anwendung
NUR ZUR INTRAVITREALEN ANWENDUNG.
ILUVIEN ist nur zur intravitrealen Anwendung bestimmt und muss von einem Facharzt für Augenheilkunde angewendet werden, der über Erfahrung mit intravitrealen Injektionen verfügt. Das Einsetzen des intravitrealen Implantats muss unter kontrollierten aseptischen Bedingungen erfolgen; dazu gehört die Verwendung steriler Handschuhe, eines sterilen Abdecktuchs und eines sterilen Lidspekulums (oder eines entsprechenden Instruments). Vor dem Einsetzen muss eine adäquate Anästhesie durchgeführt und ein Breitband-Mikrobizid angewendet werden.
1. Präoperativ können nach dem Ermessen des behandelnden Augenarztes antibiotische Augentropfen angewendet werden.
2. Unmittelbar vor dem Einsetzen wird an der Einbringungsstelle (empfohlen wird der inferotemporale Quadrant) eine Lokalanästhesie durchgeführt, entweder in Form eines Tropfens mit anschließender Anwendung eines mit Anästhetikum getränkten Wattestäbchens oder als subkonjunktivale Anwendung eines geeigneten Anästhetikums.
3. Zwei bis drei Tropfen eines geeigneten lokalen Antiseptikums werden lokal im unteren Fornix angewendet. Die Lider können mit einem Wattestäbchen abgerieben werden, das zuvor mit einem geeigneten lokalen Antiseptikum getränkt wurde. Ein steriles Lidspekulum platzieren. Den Patienten nach oben schauen lassen und ein mit einem geeigneten Antiseptikum getränktes Wattestäbchen an der Einbringungsstelle anwenden. Vor dem Einsetzen von ILUVIEN 3060 Sekunden warten, bis das lokale Antiseptikum getrocknet ist.
4. Die Außenseite der Verpackungsschale kann nicht als steril betrachtet werden. Eine Assistenzperson (nicht steril) sollte die Schale aus dem Karton herausnehmen und den Deckel von der Schale abziehen, ohne dabei die Innenfläche zu berühren. Anschließend durch das Sichtfenster des Applikatorsystems prüfen, dass sich ein Arzneimittelimplantat in der Schale befindet.
5. Den Applikator von der Schale nehmen; dabei müssen sterile Handschuhe getragen werden. Nur die sterile Oberfläche und der Applikator dürfen berührt werden.
6. Um die zusammen mit dem Implantat eingebrachte Menge an Luft so gering wie möglich zu halten, erfolgt die Anwendung in zwei Schritten. Vor dem Einsetzen der Nadel in das Auge den Knopf bis zum ersten Haltepunkt (die gerundeten schwarzen Markierungen) herunterdrücken.
Am ersten Anschlagpunkt den Knopf loslassen, so dass er in die Position „UP“ gelangt.
7. Die optimale Platzierung des Implantats ist unterhalb der Sehnervenpapille, hinter dem Äquator. Dies lässt sich erreichen, indem die Nadel auf den unteren Teil der Sehnervenpapille gerichtet wird. Mit Hilfe eines Tastzirkels einen Punkt 4 Millimeter inferotemporal vom Limbus abmessen.
8. Die Schutzkappe von der Nadel abziehen.
9. Vorsichtig die Bindehaut verschieben, damit die Einstichstellen im Konjunktival- und Skleralgewebe nach dem Herausziehen der Nadel nicht übereinander liegen. Es ist sorgfältig darauf zu achten, dass die Nadel nicht mit dem Lidrand oder den Wimpern in Kontakt kommt. Die Nadel in das Auge einführen. Zum Platzieren des Implantats den Knopf, der sich in der Position UP befindet, bis ans Ende nach vorne drücken und die Nadel herausziehen.
10. Das Lidspekulum entfernen und mittels indirekter Ophthalmoskopie prüfen, ob das Implantat richtig platziert wurde, eine adäquate Perfusion der Zentralarterie der Netzhaut vorhanden ist und keine anderen Komplikationen aufgetreten sind.
Um die erfolgreiche Platzierung des Implantats im Corpus vitreum zu überprüfen, sollte der betreffende Quadrant anschließend mit Hilfe von indirekter Ophthalmoskopie untersucht werden. Die Sichtbarkeit des Implantats kann eventuell durch leichten Druck auf die Sklera verbessert werden. Die Untersuchung sollte unmittelbar nach dem Einsetzen eine Prüfung der Perfusion des Sehnervenkopfs einschließen. Eine sofortige Messung des IOD kann nach dem Ermessen des Augenarztes ebenfalls durchgeführt werden.
Nach dem Eingriff müssen die Patienten auf potenzielle Komplikationen wie Endophthalmitis, erhöhter intraokulärer Druck, Netzhautablösung und Glaskörperblutungen oder Ablösungen überwacht werden. Zwei bis sieben Tage nach dem Einsetzen des Implantats ist eine biomikroskopische Untersuchung mit Tonometrie durchzuführen.
Es wird empfohlen, die Patienten danach mindestens vierteljährlich auf mögliche Komplikationen zu überwachen, da das Fluocinolonacetonid über einen längeren Zeitraum von etwa 36 Monaten abgegeben wird (siehe Abschnitt 4.4).
4.3 Gegenanzeigen
Bei vorbestehendem Glaukom oder bei aktiver oder vermuteter Infektion des Auges oder der Periokularregion, einschließlich der meisten Viruserkrankungen der Hornhaut oder Bindehaut, wie aktive epitheliale Herpes-simplex-Keratitis (dendritische Keratitis), Vaccinia, Varizellen, mykobakterieller Infektion und Pilzerkrankungen, ist die Anwendung eines intravitrealen Implantats mit ILUVIEN kontraindiziert.
ILUVIEN ist kontraindiziert bei Patienten mit Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Intravitreale Injektionen sind mit Endophthalmitis, erhöhtem intraokulärem Druck, Netzhautablösung und Glaskörperblutungen oder Ablösungen in Verbindung gebracht worden. Die Patienten sollten angewiesen werden, alle auf Endophthalmitis hinweisenden Symptome unverzüglich zu melden. Eine Überwachung der Patienten in den zwei bis sieben Tagen nach der Injektion kann die frühzeitige Erkennung und Behandlung von okulären Infektionen, erhöhtem intraokulärem Druck oder sonstigen Komplikationen ermöglichen. Es wird empfohlen, den intraokulären Druck danach mindestens vierteljährlich zu überwachen.
Die intravitreale Anwendung von Corticosteroiden kann Katarakt, erhöhten intraokulären Druck und Glaukom verursachen und das Risiko von Sekundärinfektionen erhöhen.
Die Unbedenklichkeit und Wirksamkeit von ILUVIEN bei der gleichzeitigen Anwendung an beiden Augen ist nicht geprüft worden. Eine gleichzeitige Behandlung beider Augen wird nicht empfohlen, solange die systemische und okuläre Reaktion des Patienten auf das erste Implantat nicht bekannt ist.
In den FAME-Studien waren Katarakt-OPs bei allen phaken Patienten in der mit ILUVIEN behandelten Gruppe etwa 3-mal häufiger (80,0 %) als in der Placebo-Gruppe (27,3 %). Die mittlere Zeitdauer bis zur Meldung von Katarakt als unerwünschtes Ereignis betrug etwa 14 Monate. Die durch Ausbildung einer Katarakt entstandenen Sehstörungen traten bei phaken Patienten vor der Kataraktextraktion etwa vom 9. bis 18. Monat nach der Implantation auf (siehe Abschnitt 4.8).
In der Gesamtpopulation der klinischen Studien, in die keine Patienten mit einem prätherapeutischen IOD > 21 mmHg aufgenommen worden waren, lag der Anteil der Patienten, die eine Behandlung mit IOD-senkenden Arzneimitteln benötigten, bei den mit ILUVIEN behandelten Patienten bei 38 %, verglichen mit 14 % in der Placebo-Gruppe. Dieser Anteil stieg bis auf 47 % bei denjenigen Patienten, deren IOD prätherapeutisch über dem Mittelwert lag (>15 mmHg). Operative Eingriffe zur Behandlung von okulärer Hypertonie waren bei 4,8 % der mit ILUVIEN behandelten Patienten erforderlich; im Vergleich dazu war dies bei 0,5 % der Placebo-behandelten Patienten der Fall (siehe Abschnitt 4.8). Daher sollte ILUVIEN bei Patienten mit einem hohen prätherapeutischen intraokulären Druck mit Vorsicht angewendet und der IOD streng überwacht werden.
Falls es zu Erhöhungen des IOD kommt, die nicht auf eine IOD-senkende Medikation oder IOD-senkende Verfahren ansprechen, kann das ILUVIEN-Implantat per Vitrektomie entfernt werden.
In den FAME-Studien wurde eine begrenzte Anzahl von Patienten mit Typ-1-Diabetes untersucht, allerdings zeigten diese Patienten keine signifikant andere Reaktion auf ILUVIEN als Patienten mit Typ-2-Diabetes.
Es gibt nur begrenzte Erfahrungen mit der Wirkung, die ILUVIEN im Anschluss an eine Vitrektomie auf die Augen hat. Nach einer Vitrektomie dürfte die Arzneimittel-Clearance wahrscheinlich beschleunigt sein, ein Einfluss auf die Konzentrationen im Fließgleichgewicht wird jedoch nicht erwartet. Die Wirkungsdauer des Implantats könnte dadurch verkürzt werden.
In den FAME-Studien wurden 24 % der Patienten in der Placebo-Gruppe zu irgendeinem Zeitpunkt entweder mit Antikoagulanzien oder mit Thrombozytenaggregationshemmern behandelt. In der mit ILUVIEN behandelten Gruppe waren es dagegen 27 % der Patienten. Bei Patienten, die begleitend zur ILUVIEN-Behandlung oder innerhalb von 30 Tagen nach Behandlungsabschluss koagulationshemmende Arzneimittel oder Thrombozytenaggregationshemmer erhalten hatten, traten Bindehauteinblutungen geringfügig häufiger auf als bei den Placebo-behandelten Patienten (0,5 % mit Placebo gegenüber 2,7 % mit ILUVIEN). Das einzige andere Ereignis, das bei den mit ILUVIEN behandelten Patienten häufiger berichtet wurde, waren Komplikationen bei der Augenoperation (0 % mit Placebo gegenüber 0,3 % mit ILUVIEN).
4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Es wurden keine Studien zur Erfassung von Wechselwirkungen durchgeführt.
4.6 Fertilität, Schwangerschaft und Stillzeit
Schwangerschaft
Es liegen keine ausreichenden Erfahrungen mit der intravitrealen Anwendung von Fluocinolonacetonid bei Schwangeren vor. Eine systemische Langzeitbehandlung mit Glucocorticoiden während der Schwangerschaft erhöht das Risiko von intrauteriner Wachstumsretardierung und
Nebenniereninsuffizienz beim Neugeborenen. Tierexperimentelle Studien haben eine teratogene Wirkung nach systemischer Anwendung gezeigt (siehe Abschnitt 5.3). Obwohl die systemische Fluocinolonacetonid-Exposition nach lokaler, intraokulärer Behandlung nur sehr gering sein dürfte, wird die Anwendung von ILUVIEN während der Schwangerschaft nicht empfohlen, sofern der potenzielle Nutzen das potenzielle Risiko für den Fetus nicht rechtfertigt.
Stillzeit
Fluocinolonacetonid wird in die Muttermilch ausgeschieden. Aufgrund der Art der Anwendung und der daraus resultierenden systemischen Spiegel werden keine Auswirkungen auf das Kind erwartet. Sofern es nicht unbedingt notwendig ist, wird ILUVIEN während der Stillzeit jedoch nicht empfohlen.
Fertilität
Es sind keine Daten zur Auswirkung auf die Fertilität verfügbar.
4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Nach der Anwendung von ILUVIEN kann die Sehfähigkeit vorübergehend vermindert sein. Daher sollten die Patienten kein Fahrzeug führen und keine Maschinen bedienen, bis die Symptome zurückgegangen sind.
4.8 Nebenwirkungen
Zusammenfassung des Sicherheitsprofils
In den klinischen FAME-Studien wurde ILUVIEN an 768 Patienten mit diabetischem Makulaödem beurteilt. Zu den am häufigsten gemeldeten unerwünschten Arzneimittelwirkungen gehörten Katarakt-OP, Katarakt und erhöhter intraokulärer Druck.
In den Studien der Phase 3 benötigten 38,4 % der mit ILUVIEN behandelten Patienten eine IOD-senkende Medikation und 4,8 % benötigten einen operativen Eingriff zur IOD-Senkung. Ähnlich war die Anwendung von IOD-senkender Medikation bei Patienten, die zwei oder mehr Behandlungen mit ILUVIEN erhalten hatten.
In den Studien der Phase 3 trat bei den mit ILUVIEN behandelten Patienten in zwei Fällen Endophthalmitis auf. Dies entspricht einer Vorkommensrate von 0,2 % (2 Fälle dividiert durch 1.022 Injektionen).
Die Mehrzahl der Patienten in den klinischen FAME-Studien erhielt lediglich ein Implantat (siehe Abschnitt 5.1), dabei sind die langfristigen Sicherheitsauswirkungen bei einem Verbleiben des nicht biologisch abbaubaren Implantats im Auge nicht bekannt. In den klinischen FAME-Studien zeigen die 3-
Jahres-Daten, dass Ereignisse wie Katarakt, erhöhter intraokulärer Druck und Flusen bei Patienten, die 2 oder mehr Implantate erhalten hatten, nur geringfügig häufiger auftraten. Es wird angenommen, dass dies eher auf die erhöhte Arzneimittel-Exposition zurückzuführen ist als auf das Implantat selbst. In vorklinischen Studien gab es außer Linsenveränderungen bei Kaninchen, die 2-4 Implantate über 24 Monate erhalten hatten, keine Hinweise auf vermehrte Sicherheitsprobleme. Das Implantat besteht aus Polyimid und ähnelt im Wesentlichen der Haptik einer Intraokularlinse. Daher wird erwartet, dass es im Auge inert bleibt.
Liste der Nebenwirkungen in Tabellenform
Die folgenden Nebenwirkungen wurden als behandlungsbedingt beurteilt und nach der folgenden Konvention klassifiziert:
sehr häufig (>1/10); häufig (>1/100 bis < 1/10); gelegentlich (>1/1.000 bis < 1/100); selten (>1/10.000 bis < 1/1.000) und sehr selten (< 1/10.000). Innerhalb jeder Häufigkeitsgruppe werden die Nebenwirkungen nach abnehmendem Schweregrad angegeben.
|
Infektionen und parasitäre Erkrankungen |
Gelegentlich: Endophthalmitis |
|
Erkrankungen des Nervensystems |
Gelegentlich: Kopfschmerz |
|
Augenerkrankungen |
Sehr häufig: Katarakt-Operation, Katarakt1, erhöhter intraokulärer Druck2, Glaskörpertrübungen (Myodesopsia) Häufig: Glaukom3, Trabekulektomie, Augenschmerzen4, Glaskörperblutung, Bindehautblutung, verschwommenes Sehen5, Glaukomoperation, Sehschärfe vermindert, Vitrektomie, Trabekuloplastie Gelegentlich: retinaler Gefäßverschluss6, Erkrankung des Nervus opticus, Makulopathie, Optikusatrophie, Bindehautulkus, Neovaskularisation der Iris, retinale Exsudate, Glaskörperdegeneration, Glaskörperabhebung, Trübung der hinteren Augenkapsel, Adhäsionen der Iris, okuläre Hyperämie, Verdünnung der Sklera, Entfernung eines abgestoßenen Implantats aus der Sklera, Augenfluss, Augenjuckreiz |
|
Verletzung, Vergiftung und durch Eingriffe bedingte Komplikationen |
Gelegentlich: Implantatabstoßung, Implantat in der Sehachse, Komplikation bei einem Eingriff, Schmerzen während eines Eingriffs |
1 Schließt die MedDRA-Bezeichnungen für Katarakt (ANE), Katarakt subkapsulär, Katarakt kortikal, Kernstar und Katarakt diabetisch ein.
2 Schließt die MedDRA-Bezeichnungen für erhöhten intraokulären Druck und Augeninnendruck erhöht ein.
3 Schließt die MedDRA-Bezeichnungen für Glaukom, Weitwinkelglaukom, Borderline-Glaukom, Exkavation des Nervus opticus und Exkavation der Sehnervenpapille erhöht ein.
4 Schließt die MedDRA-Bezeichnungen für Augenschmerzen, Augenreizung und Augenbeschwerden ein.
5 Schließt die MedDRA-Bezeichnungen für verschwommenes Sehen und Sehverschlechterung ein.
6 Schließt die MedDRA-Bezeichnungen für Netzhautvenenverschluss, Verschluss einer Netzhautarterie und retinalen Gefäßverschluss ein.
Beschreibung ausgewählter Nebenwirkungen
Die Langzeitanwendung von Corticosteroiden kann Katarakt und erhöhten intraokulären Druck verursachen. Die unten angegebenen Häufigkeiten sind anhand der Befunde aller an den FAME-Studien teilnehmenden Patienten erhoben worden. Die bei Patienten mit chronischem DMÖ beobachteten Häufigkeiten unterschieden sich nicht signifikant von den in der Gesamtpopulation beobachteten.
In den klinischen Studien der Phase 3 betrug die Auftretenshäufigkeit von Katarakt bei phaken Patienten etwa 82 % bei den mit ILUVIEN behandelten Patienten und 50 % bei den Placebo-behandelten Patienten. Bei 80 % der mit ILUVIEN behandelten phaken Patienten war bis zum Jahr 3 eine Katarakt-OP erforderlich, im Vergleich dazu lag die Quote bei den Placebo-behandelten Patienten bei 27 %, wobei die meisten Patienten bis zum Monat 21 einen Eingriff benötigten. Der häufigste Katarakt-Typ in Verbindung mit Corticosteroiden ist die posteriore subkapsuläre Katarakt. Operative Eingriffe bei diesem Typ sind schwieriger und gehen möglicherweise mit einem erhöhten Komplikationsrisiko einher.
Patienten mit einem prätherapeutischen IOD > 21 mmHg wurden nicht in die FAME-Studien aufgenommen. Die Vorkommenshäufigkeit von erhöhtem intraokulärem Druck lag bei 37 %. Eine IOD-senkende Medikation benötigten 38 % der Patienten, davon benötigte die Hälfte mindestens zwei Medikationen zur Kontrolle des IOD. Ähnlich war der Einsatz von IOD-senkenden Arzneimitteln bei den Patienten, die während der Studie eine erneute Behandlung mit einem zusätzlichen Implantat erhalten hatten. Von den Patienten, die ein Implantat erhalten hatten, benötigten außerdem 5,6 % (21/375) einen chirurgischen Eingriff oder eine Laserbehandlung zur Kontrolle des IOD (Trabekuloplastik 5 (1,3 %), Trabekulektomie 10 (2,7 %), endoskopische Zykloablation 2 (0,5 %) und andere chirurgische Verfahren 6 (1,6 %)).
In der Untergruppe der Patienten mit einem über dem Mittelwert liegenden prätherapeutischen IOD (>15 mmHg) benötigten 47 % IOD-senkende Arzneimittel. Außerdem stieg in dieser Untergruppe der Anteil der chirurgischen oder lasergestützten Eingriffe auf 7,1 %. In dieser Untergruppe erhielten 5 Patienten (2,2 %) eine Trabekuloplastik, 7 (3,1 %) eine Trabekulektomie, 2 (0,9 %) eine endoskopische Zykloablation und bei 4 (1,8 %) wurden andere chirurgische Verfahren zur Glaukombehandlung angewendet.
4.9 Überdosierung
Es wurden keine Fälle von Überdosierung berichtet.
5. PHARMAKOLOGISCHE EIGENSCHAFTEN
5.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: ANTIPHLOGISTIKA, Corticosteroide, rein ATC-Code: S01BA15
Corticosteroide hemmen die Entzündungsreaktion auf eine Vielzahl von Erregern. Sie hemmen das Auftreten von Ödemen, Fibrinablagerungen, Kapillarerweiterung, Leukozytenmigration,
Kapillarproliferation, Fibroblastenproliferation, Kollagenablagerung und Narbenbildung in Verbindung mit einer Entzündung.
Corticosteroide wirken vermutlich durch die Induktion von Proteinen, die hemmend auf die Aktivität der Phospholipase A wirken und als Lipocortine bezeichnet werden. Man geht davon aus, dass diese Proteine die Biosynthese potenter Entzündungsmediatoren wie Prostaglandine und Leukotriene dadurch kontrollieren, dass sie die Freisetzung der gemeinsamen Vorstufe Arachidonsäure hemmen. Arachidonsäure wird mit Hilfe der Phospholipase A2 aus Membranphospholipiden freigesetzt. Es konnte auch gezeigt werden, dass Corticosteroide die Spiegel des vaskulären endothelialen Wachstumsfaktors senken, eines Proteins, das die vaskuläre Permeabilität erhöht und Ödeme verursacht.
Die Wirksamkeit von ILUVIEN wurde in zwei randomisierten, multizentrischen, doppelt verblindeten Parallelstudien beurteilt. In diese Studien, die jeweils eine Nachuntersuchungsphase von 3 Jahren einschlossen, wurden Patienten mit diabetischem Makulaödem aufgenommen, die zuvor mindestens einmal mit Laserphotokoagulation behandelt worden waren. Von den Patienten erhielten 74,4 % jeweils 1 Implantat, 21,6 % erhielten 2 Implantate, 3,5 % erhielten 3 Implantate und 0,5 % erhielten 4 Implantate sowie 0 % > 4 Implantate). Der primäre Wirksamkeitsendpunkt beider Studien war der Anteil der Patienten, deren Visus sich nach 24 Monaten um 15 Buchstaben oder mehr verbessert hatte. In beiden Studien wurde der primäre Endpunkt für ILUVIEN erreicht (siehe Abbildung 1 mit den zusammenfassenden Ergebnissen für den primären Wirksamkeitsendpunkt).
Abbildung 1: Prozentsatz der Patienten mit einer gegenüber dem prätherapeutischen Ausgangswert erzielten Verbesserung von >15 Buchstaben (Zusammenfassung der FAME-Studien)
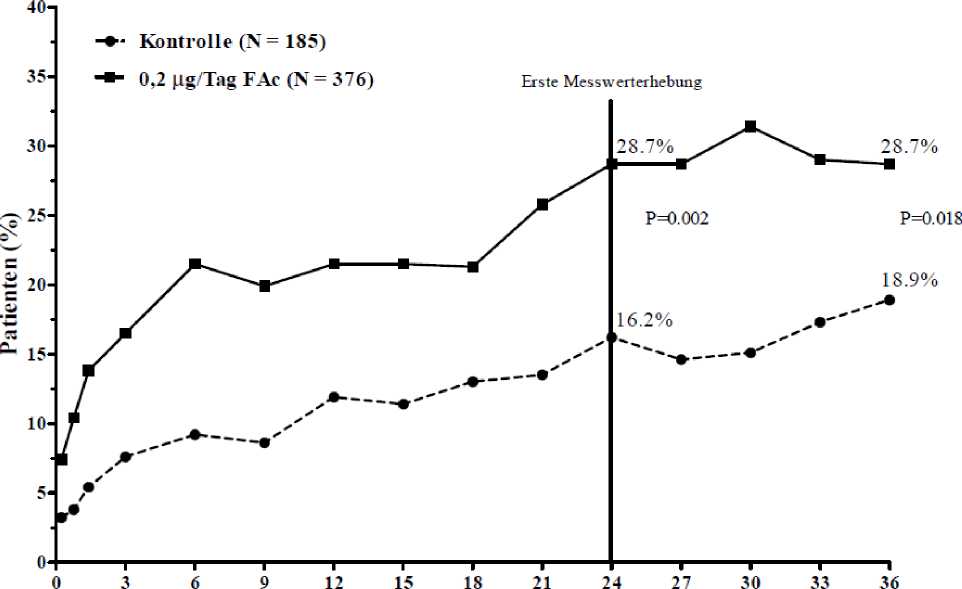
Monate
Wenn die Wirksamkeit abhängig von der Dauer der Erkrankung beurteilt wurde, zeigten die Patienten mit einer über dem Mittelwert (>3 Jahre) liegenden Dauer des DMÖ ein signifikant günstiges Ansprechen auf ILUVIEN, während sich bei den Patienten mit weniger lang anhaltendem DMÖ in Bezug auf eine Visusverbesserung kein zusätzlicher Nutzen gegenüber der Kontrollbehandlung zeigte (Abbildungen 2 und 3). Diese Untergruppendaten stützen die im Abschnitt 4.1. angegebene Indikation der Anwendung bei Patienten mit chronischem DMÖ (d.h. eine Dauer von mindestens 3 Jahren).
Abbildung 2: Vergleich des Prozentsatzes der Patienten mit einer gegenüber der prätherapeutischen BCVA (bestkorrigierten Sehschärfe) erreichten Verbesserung von >15 Buchstaben und Durchschnittliche Veränderung der Zunahme der fovealen Netzhautdicke nach der Dauer in der DMÖ-Untergruppe >3 Jahre, verglichen mit dem prätherapeutischen Wert
Krste Ales swerterhe bims
Kontrolle (N = 112)
p < 0.001
p < 0.001
0,2 usT.ig FAc 1> = 209)
34.4%
34.0%
20-
15-
♦---♦
13.4%
13.4%
Monate
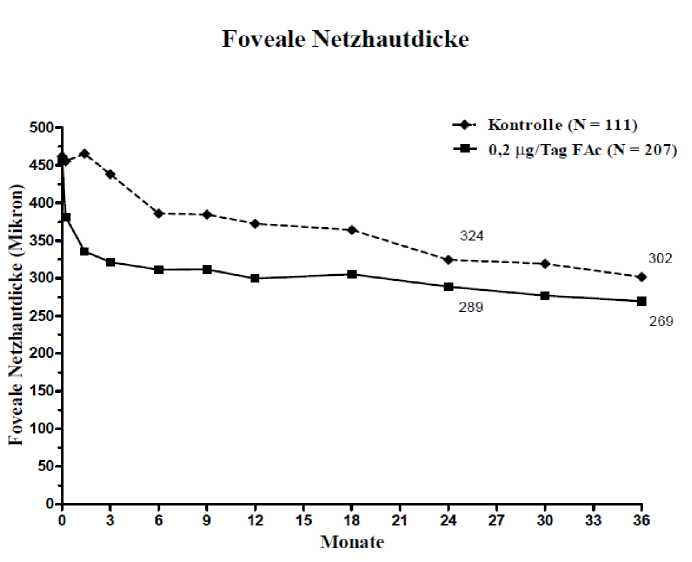
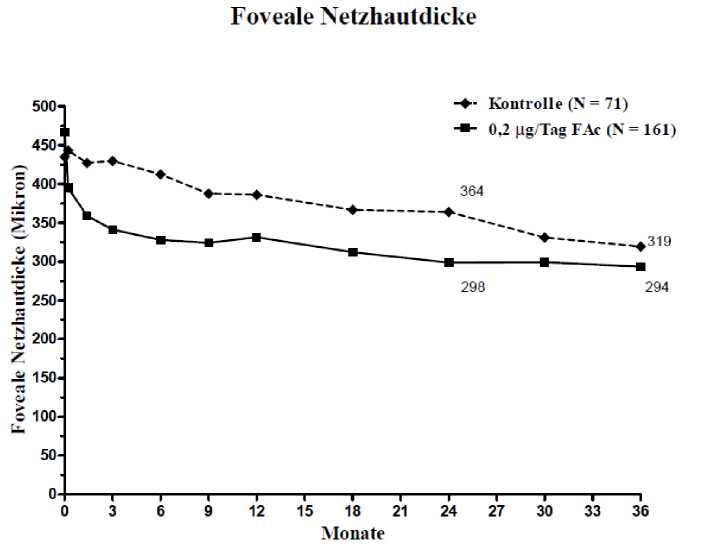
Abbildung 3: Vergleich der durchschnittlichen Veränderung der Zunahme der fovealen Netzhautdicke im Vergleich zum prätherapeutischen Wert und Prozentsatz der Patienten mit einer gegenüber der prätherapeutischen BCVA (bestkorrigierten Sehschärfe) erreichten Verbesserung von >15 Buchstaben nach der Dauer in der DMÖ-Untergruppe < 3 Jahre
Prozentsatz der Patienten mit Ansprechen > 15 Buchstaben
40
Ei ste Messwerte rhebuug
■ Kontrolle (> — 72)
35
30
i' 25
15
0
0,2 iig Tag FAc (> = 166)
p = 0.934
p = 0.275
27.8%
21.7%
20.8%
22.3%
Mouate
Die Europäische Arzneimittelagentur hat für alle Untergruppen der pädiatrischen Population auf die Verpflichtung zur Vorlage von Studienergebnissen mit ILUVIEN zur Behandlung von diabetischem Makulaödem verzichtet. Siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen.
5.2 Pharmakokinetische Eigenschaften
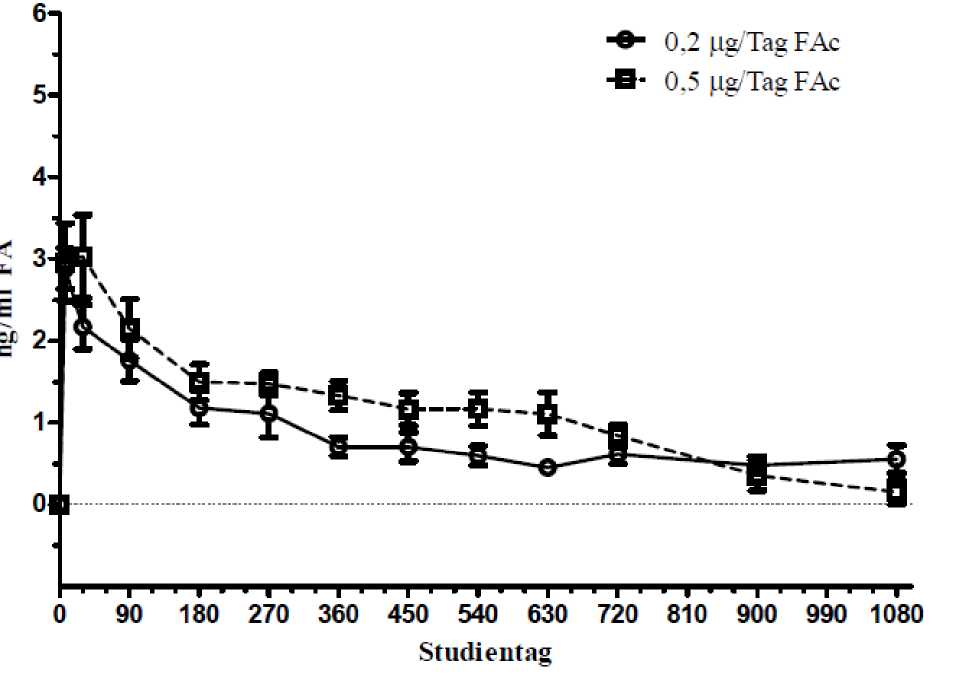
In einer Studie zur Pharmakokinetik beim Menschen (C-01-06-002, die so genannte FAMOUS-Study) lagen die Konzentrationen von Fluocinolonacetonid im Plasma zu allen Zeitpunkten von Tag 1 bis Monat 36 unterhalb der unteren Nachweisgrenze des Assays (100 pg/ml), was auf eine nicht nennenswerte systemische Exposition hinweist. Die Höchstkonzentrationen von Fluocinolonacetonid im Kammerwasser wurden bei den meisten Patienten am Tag 7 beobachtet. Die Fluocinolonacetonid-Konzentrationen im Kammerwasser sanken im Laufe der ersten 3-6 Monate und blieben bei nicht erneut behandelten Patienten bis zum Monat 36 im Wesentlichen gleich. Bei den erneut behandelten Patienten trat eine zweite Fluocinolonacetonid-Spitzenkonzentration auf, die der im Anschluss an die Initialdosis beobachteten glich. Nach einer erneuten Behandlung gingen die Kammerwasser-Konzentrationen von Fluocinolonacetonid auf Werte zurück, die in etwa den während der Erstbehandlung beobachteten entsprachen.
Abbildung 4: FA-Spiegel im Kammerwasser menschlicher Patienten, die 1 Implantat erhalten hatten (FAMOUS-Studie)
5.3 Präklinische Daten zur Sicherheit
Präklinische Effekte wurden nur nach Expositionen beobachtet, die ausreichend über der maximalen humantherapeutischen Exposition lagen. Die Relevanz für den Menschen wird als gering bewertet.
Daten zur Mutagenität, Kanzerogenität sowie zur Reproduktions- oder Entwicklungstoxizität sind für ILUVIEN nicht verfügbar. Bei Mäusen und Kaninchen hat sich Fluocinolonacetonid nach systemischer Anwendung als teratogen erwiesen.
Beurteilung der Risiken für die Umwelt (Environmental Risk Assessment IERAI)
Angesichts der extrem niedrigen Dosis des implantierten Fluocinolonacetonids besteht bei verordnungsgemäßer Anwendung des ILUVIEN-Implantats kein nennenswertes Risiko für die Umwelt.
Der speziell konzipierte Injektionsapplikator für ILUVIEN stellt kein Risiko für die Umwelt dar, da er nach einmaligem Gebrauch unverzüglich in einer Abwurfbox für medizinisches/chirurgisches Material entsorgt wird.
Hinweise zur Beseitigung und Handhabung siehe Abschnitt 6.6.
6. PHARMAZEUTISCHE ANGABEN
6.1 Liste der sonstigen Bestandteile
Poly(vinylalkohol)
Polyimid-Röhrchen
Silikonklebstoff
6.2 Inkompatibilitäten
Nicht zutreffend.
6.3 Dauer der Haltbarkeit
2 Jahre.
Nach dem Öffnen des Deckels sofort verwenden.
6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Nicht über 30°C lagern. Nicht im Kühlschrank lagern oder einfrieren. Die verschlossene Schale erst unmittelbar vor der Anwendung öffnen.
6.5 Art und Inhalt des Behältnisses
Das Implantat wird in einem Applikator zum einmaligen Gebrauch mit einer Injektionsnadel 25 G geliefert. Jeder sterile Applikator enthält ein hellbraunes, 3,5 mm langes zylindrisches Implantat. Der Applikator ist in einer Kunststoffschale mit Verschlussdeckel verpackt.
6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung
Applikator in einer Abwurfbox für infektiöses Material sicher entsorgen.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
7. INHABER DER ZULASSUNG
Alimera Sciences Limited Centaur House Ancells Road Fleet
Hampshire GU51 2UJ Vereinigtes Königreich
Tel.: 069 222 23387 Tel.: 0800 664 6695
8. ZULASSUNGSNUMMER
82809.00.00
9. DATUM DER ERTEILUNG DER ZULASSUNG / VERLÄNGERUNG DER ZULASSUNG Juli 2012
10. STAND DER INFORMATION
12/2013
11. VERKAUFSABGRENZUNG
Verschreibungspflichtig
Dieses Arzneimittel enthält einen Stoff, dessen Wirkung zur Behandlung von Sehstörungen in Verbindung mit chronischem diabetischem Makulaödem, das auf verfügbare Therapien nur unzureichend anspricht, in der medizinischen Wissenschaft noch nicht allgemein bekannt ist.