Immunine 1200 I.e.
FACHINFORMATION
(Zusammenfassung der Merkmale des Arzneimittels)
1. BEZEICHNUNG DES ARZNEIMITTELS
IMMUNINE 600 I.E. / 1200 I.E.
Pulver und Lösungsmittel zur Herstellung einer Injektions- oder Infusionslösung
2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
Wirkstoff: Blutgerinnungsfaktor IX vom Menschen
Jede Durchstechflasche mit Pulver zur Herstellung einer Injektionslösung enthält nominell 600 I.E. (IMMUNINE 600 I.E.) oder 1200 I.E. (IMMUNINE 1200 I.E.) Blutgerinnungsfaktor IX vom Menschen.
1 ml Lösung IMMUNINE enthält nach Rekonstitution in 5 ml (IMMUNINE 600 I.E.) oder 10 ml (IMMUNINE 1200 I.E.) sterilisiertem Wasser für Injektionszwecke ca. 120 I.E. Blutgerinnungsfaktor IX vom Menschen.
Die Aktivität (I.E.) von Faktor IX wurde mit Hilfe des in der Europäischen Pharmakopoe beschriebenen Einstufen-Gerinnungstests bestimmt.
Hergestellt aus Plasma humaner Spender.
Die spezifische Aktivität von IMMUNINE ist > 50 I.E. Faktor IX/mg Protein.
Vollständige Auflistung der sonstigen Bestandteile siehe Abschnitt 6.1
3. DARREICHUNGSFORM
Pulver und Lösungsmittel zur Herstellung einer Injektions- oder Infusionslösung. Weiße bis hellgelbe, gefriergetrocknete, pulvrige oder kompakte Trockensubstanz.
4. KLINISCHE ANGABEN
4.1 Anwendungsgebiete
Therapie und Prophylaxe von Blutungen bei Patienten mit Hämophilie B (angeborener Faktor-IXMangel).
IMMUNINE ist für die Anwendung in allen Altersgruppen - bei Kindern älter als 6 Jahre bis hin zu Erwachsenen - indiziert.
Die Anwendung von IMMUNINE bei Kindern unter 6 Jahren kann nicht empfohlen werden, da hierzu nur unzureichende Daten vorliegen.
4.2 Dosierung und Art der Anwendung
Die Behandlung sollte unter der Aufsicht eines mit der Therapie der Hämophilie erfahrenen Arztes eingeleitet werden.
Dosierung
Dosierung und Dauer der Substitutionstherapie richten sich nach dem Schweregrad des Faktor-IXMangels, dem Ort und Ausmaß der Blutung und dem klinischen Zustand des Patienten.
Die verabreichten Faktor-IX-Einheiten werden in Internationalen Einheiten (I.E.) angegeben, die vom aktuellen WHO-Standard für Faktor-IX-Produkte abgeleitet sind. Die Faktor-IX-Aktivität im Plasma wird entweder als Prozentsatz (relativ zum normalen menschlichen Plasma) oder in Internationalen Einheiten (relativ zum internationalen Standard für Faktor-IX-Konzentrate im Plasma) angegeben. Eine Internationale Einheit (I.E.) Faktor-IX-Aktivität entspricht der Menge an Faktor IX in 1 ml normalem menschlichen Plasma.
Bedarfsbehandlung
Die Berechnung der erforderlichen Faktor-IX-Dosis basiert auf der empirischen Erkenntnis, dass 1 Internationale Einheit (I.E.) Faktor IX pro kg Körpergewicht bei Patienten ab 12 Jahren die Faktor-IX-Aktivität im Plasma um ca. 1,1 % der normalen Aktivität erhöht.
Die benötigte Dosis wird mit folgender Formel berechnet:
Benötigte Einheiten = Körpergewicht (kg) x gewünschter Faktor-IX-Anstieg (%) (IE/dl) x 0,9
Dosis und Häufigkeit der Verabreichung sollten sich immer nach der klinischen Wirksamkeit des Produktes im Einzelfall richten. Faktor-IX-Produkte müssen nur selten mehr als 1 x pro Tag verabreicht werden.
Bei folgenden hämorrhagischen Ereignissen darf die Faktor-IX-Aktivität im entsprechenden Zeitraum nicht unter den angegebenen Plasmaspiegel (in % der Norm oder in I.E./dl) sinken.
Die folgende Tabelle enthält Richtwerte für die Dosierung bei Blutungen und chirurgischen Eingriffen:
|
Grad der Blutung / Art des chirurgischen Eingriffs |
Erforderlicher Faktor-IX-Plasmaspiegel (% des Normalwertes) (I.E./dl) |
Häufigkeit der Dosierung (Stunden) / Behandlungsdauer (Tage) |
|
Blutung | ||
|
Frühstadium von Gelenks- oder Muskelblutung, oder Blutung in der Mundhöhle |
20 - 40 |
Alle 24 Stunden wiederholen. Mindestens 1 Tag, bis die Blutung (angezeigt durch Schmerzen), zum Stehen gekommen, oder die Wundheilung erreicht ist. |
|
Ausgeprägtere Gelenkblutung, Muskelblutung oder Hämatom |
30 - 60 |
Infusion alle 24 Stunden wiederholen; für normalerweise 3 - 4 Tage oder länger, bis die Schmerzen und die akute Beeinträchtigung beseitigt sind. |
|
Lebensbedrohliche Blutungen wie bei Eingriffen am Kopf, Blutungen in den Hals, starken Blutungen im Abdomen |
60 - 100 |
Infusion alle 8 - 24 Stunden wiederholen, bis die Gefahr für den Patienten vorüber ist. |
|
Chirurgische Eingriffe | ||
|
Kleinere Eingriffe einschließlich Zahnextraktion |
30 - 60 |
Alle 24 Stunden; mindestens 1 Tag, bis die Wundheilung erreicht ist. |
|
Größere Eingriffe |
80 - 100 (prä- und postoperativ) |
Die Infusion alle 8 - 24 Stunden wiederholen, bis eine adäquate Wundheilung erreicht ist; anschließend eine Behandlungsdauer von mindestens 7 weiteren Tagen, um eine Faktor IX Aktivität von 30 - 60 % (I.E./dl) aufrecht zu erhalten. |
Zur Langzeitprophylaxe von Blutungen bei Patienten mit schwerer Hämophilie B liegen die üblichen Dosen zwischen 20 und 40 I.E. FIX/kg im Abstand von 3 - 4 Tagen.
In manchen Fällen, besonders bei jüngeren Patienten, können kürzere Dosierungsabstände oder höhere Dosen erforderlich sein.
Es wird empfohlen, den Faktor IX-Plasmaspiegel während des Behandlungsverlaufs in geeigneter Weise zu überwachen, damit die zu verabreichende Dosis und die Häufigkeit wiederholter Infusionen festgelegt werden können. Besonders bei größeren chirurgischen Eingriffen ist eine genaue Überwachung der Substitutionstherapie durch Bestimmung des Blutgerinnungsstatus (Faktor-IXAktivität im Plasma) unerlässlich. Patienten können auf die Faktor IX-Gabe unterschiedlich ansprechen. Dies zeigt sich in unterschiedlichen Werten der in-vivo-Recovery und unterschiedlichen Halbwertzeiten.
Kinder und Jugendliche
Zurzeit vorliegende Daten werden in Abschnitt 5.2 beschrieben; eine Dosierungsempfehlung kann jedoch nicht gegeben werden.
Art der Anwendung
Intravenöse Verabreichung. Die Infusionsgeschwindigkeit sollte nicht mehr als 2 ml pro Minute betragen.
Hinweise zur Rekonstitution des Arzneimittels vor der Anwendung, siehe Abschnitt 6.6.
4.3 Gegenanzeigen
• Überempfindlichkeit gegen den Wirkstoff oder einen der sonstigen Bestandteile.
• Disseminierte Intravasale Gerinnung (DIC) und/oder Hyperfibrinolyse.
• Bekannte Heparin-Allergie oder Heparin-induzierte Thrombopenie in der Anamnese.
Nach entsprechender Abklärung dieser Erkrankungen darf IMMUNINE nur bei lebensbedrohlichen Blutungen verabreicht werden.
4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Überempfindlichkeit
Unter der Anwendung von IMMUNINE können allergische Überempfindlichkeitsreaktionen auftreten. Das Produkt enthält außer Faktor IX Spuren anderer menschlicher Plasmaproteine. Die Patienten sind anzuweisen, beim Auftreten von Symptomen einer Überempfindlichkeitsreaktion die Behandlung sofort abzubrechen und ihren Arzt zu kontaktieren. Die Patienten und/oder ihre Betreuer sind über die frühen Anzeichen einer Überempfindlichkeitsreaktion wie Nesselausschlag, generalisierte Urtikaria, Engegefühl in der Brust, Stenoseatmung, Hypotonie und Anaphylaxie aufzuklären. Im Falle eines Schocks ist eine Schocktherapie nach aktuellem medizinischem Standard durchzuführen.
Inhibitoren
Nach wiederholter Gabe humaner Blutgerinnungsfaktor-IX-Produkte sollten die Patienten auf die Bildung neutralisierender Antikörper (Inhibitoren) hin überwacht werden. Inhibitoren sind unter Verwendung geeigneter biologischer Tests in Bethesda-Einheiten (B.E.) zu quantifizieren.
Falls die erwarteten Faktor-IX-Plasmaaktivitäten nicht erreicht werden oder die Blutung mit einer angemessenen Dosis nicht beherrscht werden kann, muss ein Test zur Bestimmung von Faktor-IXInhibitoren durchgeführt werden. Bei Patienten mit hohen Inhibitorwerten ist die Faktor-IX-Therapie möglicherweise nicht wirksam, so dass andere therapeutische Maßnahmen erwogen werden müssen. Die Behandlung dieser Patienten sollte unter der Anleitung von Ärzten erfolgen, die über Erfahrung in
der Behandlung von Patienten mit Hämophilie verfügen. Aus diesem Grund sollte ein spezialisiertes Hämophilie-Zentrum kontaktiert werden.
In der Literatur wird von einem Zusammenhang zwischen dem Auftreten von Faktor-IX-Inhibitoren und allergischen Reaktionen berichtet. Es ist zu beachten, dass bei Patienten mit Faktor-IX-Inhibitoren bei erneuter Exposition gegenüber Faktor IX ein erhöhtes Risiko für das Auftreten einer Anaphylaxie besteht.
Wegen des Risikos allergischer Reaktionen auf Faktor-IX-Produkte sollten die ersten Faktor-IXGaben nach Ermessen des behandelnden Arztes unter medizinischer Aufsicht erfolgen, so dass eine mögliche allergische Reaktion angemessen behandelt werden kann.
Thromboembolie, DIC, Fibrinolyse
Da bei Verwendung von Faktor-IX-Konzentraten in der Vergangenheit thromboembolische Komplikationen beobachtet wurden, wobei das Risiko bei weniger gut gereinigten Produkten höher ist, kann die Gabe von Faktor-IX-Produkten für Patienten mit Zeichen einer Fibrinolyse oder einer Disseminierten Intravasalen Gerinnung (DIC) eine potentielle Gefährdung darstellen. Wegen des möglichen Risikos thromboembolischer Komplikationen ist bei Patienten mit Lebererkrankungen, Thrombophilie, Hyperkoagulabilität, Angina pectoris, einer Koronarerkrankung oder einem akuten Myokardinfarkt sowie bei postoperativen Patienten, Frühgeborenen, Neugeborenen oder bei Patienten, bei denen das Risiko für eine Thrombose oder Verbrauchskoagulopathie besteht, eine klinische Beobachtung, einschließlich geeigneter Testverfahren, erforderlich, um Frühzeichen einer thromboembolischen Komplikation oder einer Verbrauchskoagulopathie festzustellen. In den genannten Fällen muss der Nutzen der Behandlung mit IMMUNINE gegen das mögliche Risiko dieser Komplikationen abgewogen werden.
Bei Verdacht auf DIC ist die Substitutionstherapie mit IMMUNINE unverzüglich zu beenden. Virussicherheit
• Standardmaßnahmen zur Vorbeugung von Infektionen, die sich durch den Einsatz von Arzneimitteln ergeben, die aus Humanblut oder Humanplasma hergestellt sind, schließen die Auswahl der Spender und das Screening der einzelnen Spenden und Plasmapools auf spezifische Infektionsmarker sowie effektive Schritte zur Inaktivierung/Entfernung von Viren im Herstellungsverfahren ein. Dennoch kann bei der Verabreichung von Arzneimitteln aus Humanblut oder Humanplasma die Möglichkeit der Übertragung von Infektionserregern nicht völlig ausgeschlossen werden. Dies gilt auch für bislang unbekannte oder neu aufgetretene Viren und andere Pathogene.
• Die getroffenen Maßnahmen werden als wirksam erachtet für umhüllte Viren wie das humane Immunschwächevirus (HIV), das Hepatitis-B-Virus (HBV), das Hepatitis-C-Virus (HCV) und für das nicht-umhüllte Hepatitis-A-Virus (HAV).
• Die getroffenen Maßnahmen sind für nicht-umhüllte Viren wie das Parvovirus B19 möglicherweise nur begrenzt wirksam. Eine Infektion mit dem Parvovirus B19 kann schwerwiegende Folgen bei Schwangeren (fetale Infektion) und bei Menschen mit Immunschwäche oder gesteigertem Erythrozytenumsatz (z. B. bei hämolytischer Anämie) haben.
• Wird ein aus menschlichem Plasma hergestelltes Faktor-IX-Konzentrat regelmäßig oder wiederholt verabreicht, müssen geeignete Impfungen (Hepatitis A und B) in Betracht gezogen werden.
Vorsichtsmaßnahmen bei der Anwendung
Natriumgehalt
IMMUNINE 600 I.E. enthält 20 mg Natrium pro Durchstechflasche (berechneter Wert).
IMMUNINE 1200 I.E. enthält 41 mg Natrium pro Durchstechflasche (berechneter Wert).
Dies ist bei Patienten unter natriumarmer Diät zu berücksichtigen.
Es wird dringend empfohlen, jede Verabreichung von IMMUNINE mit Produktnamen und Chargenbezeichnung zu dokumentieren, um die Verbindung zwischen Patient und verwendeter Produktcharge herstellen zu können.
4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Es wurden keine Wechselwirkungsstudien mit IMMUNINE durchgeführt.
4.6 Fertilität, Schwangerschaft und Stillzeit
Reproduktionsstudien an Tieren wurden mit Faktor IX nicht durchgeführt. Aufgrund des seltenen Auftretens der Hämophilie B bei Frauen liegen über die Anwendung von Faktor-IX-Konzentraten während der Schwangerschaft und Stillzeit keine Erfahrungen vor. IMMUNINE darf daher in der Schwangerschaft und Stillzeit nur bei eindeutiger Indikationsstellung angewendet werden.
Die Auswirkungen von IMMUNINE auf die Fertilität wurden nicht untersucht.
Bezüglich des Risikos einer Infektion mit dem Parvovirus B19 lesen Sie bitte in Abschnitt 4.4 den Warnhinweis unter der Überschrift „Virussicherheit“.
4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Es wurden keine Auswirkungen auf die Verkehrstüchtigkeit oder die Fähigkeit zum Bedienen von Maschinen beobachtet.
4.8 Nebenwirkungen
Zusammenfassung des Sicherheitsprofils
Überempfindlichkeitsreaktionen oder allergische Reaktionen (u. a. Angioödem, Brennen oder Stechen an der Infusionsstelle, Schüttelfrost, Flush, generalisierte Urtikaria, Kopfschmerzen, Nesselsucht, Hypotonie, Lethargie, Übelkeit, Ruhelosigkeit, Tachykardie, Engegefühl in der Brust, Kribbeln, Erbrechen, Stenoseatmung) wurden in eher seltenen Fällen bei Patienten, die mit Faktor-IX-haltigen Produkten behandelt wurden, beobachtet.
In manchen Fällen entwickelten sich diese Reaktionen zu einer schweren Anaphylaxie. Diese Reaktionen traten in enger zeitlicher Verbindung mit der Bildung von Faktor-IX-Inhibitoren auf (siehe auch Abschnitt 4.4).
Bei Hämophilie B-Patienten mit Faktor-IX-Inhibitoren und allergischen Reaktionen in der Anamnese wurde über das Auftreten eines nephrotischen Syndroms nach versuchter Immuntoleranzinduktion berichtet.
In seltenen Fällen wurde Fieber beobachtet.
Patienten mit Hämophilie B können neutralisierende Antikörper (Inhibitoren) gegen Faktor IX bilden (siehe Abschnitt 4.4). Falls solche Inhibitoren auftreten, manifestiert sich dieser Zustand als eine unzureichende klinische Antwort. Es wird empfohlen, in diesen Fällen ein spezialisiertes HämophilieZentrum zu kontaktieren.
Es besteht ein potenzielles Risiko für das Auftreten thromboembolischer Ereignisse nach der Anwendung von Faktor-IX-Produkten, wobei das Risiko bei Präparaten mit geringerer Reinheit höher ist. Die Anwendung von weniger reinen Faktor-IX-Produkten wurde mit Fällen von Myokardinfarkt, Disseminierter Intravasaler Gerinnung, Venenthrombose und Lungenembolie in Zusammenhang gebracht. Hochgereinigte Faktor-IX-Produkte wurden mit solchen Nebenwirkungen nur selten in Verbindung gebracht.
Zur Virussicherheit siehe Abschnitt 4.4.
Tabellarische Zusammenfassung der Nebenwirkungen
Die nachstehende Tabelle entspricht den Systemorganklassen gemäß MedDRA-Datenbank (Angaben nach SOC und bevorzugtem Terminus).
Die Liste der Nebenwirkungen beruht auf den Meldungen aus sechs klinischen Studien, die mit IMMUNINE bei 197 Patienten durchgeführt worden sind, sowie auf Spontanmeldungen im Rahmen der Pharmakovigilanz nach der Zulassung.
Die Häufigkeitsangaben entsprechen der folgenden Konvention: Sehr häufig (>1/10), häufig (>1/100; <1/10), gelegentlich (>1/1.000; <1/100), selten (>1/10.000; <1/1.000), sehr selten (<1/10.000), nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
|
Systemorganklasse gemäß MedDRA-Standard |
Nebenwirkung |
Häufigkeit |
|
Erkrankungen des Blutes und des Lymphsystems |
F aktor-IX-Hemmung |
nicht bekannt |
|
Disseminierte Intravasale Gerinnung |
nicht bekannt | |
|
Erkrankungen des Immunsystems |
Allergische Reaktion |
nicht bekannt |
|
Anaphylaktische Reaktionen/ Anaphylaktoide Reaktionen |
nicht bekannt | |
|
Angioödem |
nicht bekannt | |
|
Nesselsucht |
nicht bekannt | |
|
Bei Vorliesen von Inhibitoren: Serumkrankheit |
nicht bekannt | |
|
Überempfindlichkeitsreaktion |
nicht bekannt | |
|
Erkrankungen des Nervensystems |
Kopfschmerzen |
nicht bekannt |
|
Unruhe |
nicht bekannt | |
|
Kribbeln |
nicht bekannt | |
|
Herzerkrankungen |
Myokardinfarkt |
nicht bekannt |
|
Tachykardie |
nicht bekannt | |
|
Gefäßerkrankungen |
Hypotonie |
nicht bekannt |
|
Thromboembolische Ereignisse (z. B. Lungenembolie, Venenthrombose, arterielle Thrombose, Zerebralarterienthrombose) |
nicht bekannt | |
|
Hautrötung (Flush) |
nicht bekannt | |
|
Erkrankungen der Atemwege, des Brustraums und des Mediastinums |
Rachenreizung |
gelegentlich |
|
Schmerzen im Mund- und Rachenraum |
gelegentlich | |
|
Husten (trocken) |
gelegentlich | |
|
Stenoseatmung |
nicht bekannt | |
|
Dyspnoe |
nicht bekannt | |
|
Erkrankungen des Gastrointestinaltrakts |
Übelkeit |
nicht bekannt |
|
Erbrechen |
nicht bekannt | |
|
Erkrankungen der Haut und des Unterhautzellgewebes |
Ausschlag |
gelegentlich |
|
Juckreiz |
gelegentlich | |
|
Urtikaria |
nicht bekannt | |
|
Erkrankungen der Nieren und Harnwege |
Nephrotisches Syndrom1 |
nicht bekannt |
|
Systemorganklasse gemäß MedDRA-Standard |
Nebenwirkung |
Häufigkeit |
|
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort |
Pyrexie |
gelegentlich |
|
Schüttelfrost |
nicht bekannt | |
|
Brennen und Stechen an der Infusionsstelle |
nicht bekannt | |
|
Lethargie |
nicht bekannt | |
|
Engegefühl in der Brust |
nicht bekannt |
Inhibitoren gegen Faktor IX
In klinischen Studien mit IMMUNINE wurden keine Inhibitoren gegen Faktor IX festgestellt. Es wurden keine zuvor unbehandelten Patienten (PUPs) in die klinischen Studien mit IMMUNINE aufgenommen.
Mögliche unerwünschte Wirkungen mit BlutgerinnungsFaktor-IX-Konzentraten vom Menschen: Parästhesie
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel, Paul-Ehrlich-Institut, Paul-Ehrlich-Str. 51-59, 63225 Langen, Tel: +49 6103 77 0, Fax: +49 6103 77 1234, Website: www.pei.de anzuzeigen.
4.9 Überdosierung
Es wurden keine Symptome von Überdosierung mit Blutgerinnungsfaktor IX vom Menschen berichtet.
5. PHARMAKOLOGISCHE EIGENSCHAFTEN
5.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Antihämorrhagika, Blutgerinnungsfaktor IX; ATC-Code: B02BD04
Faktor IX ist ein Einzelketten-Glykoprotein mit einem Molekulargewicht von ca. 68.000 Dalton. Es ist ein Vitamin K-abhängiger Blutgerinnungsfaktor und wird in der Leber synthetisiert. Faktor IX wird durch Faktor XIa über den intrinsischen und durch den Faktor VII/Tissue-Faktor-Komplex über den extrinsischen Weg der Blutgerinnung aktiviert. Aktivierter Faktor IX aktiviert zusammen mit aktiviertem Faktor VIII den Faktor X. Der aktivierte Faktor X wandelt Prothrombin in Thrombin um; Thrombin wandelt wiederum Fibrinogen zu Fibrin um, wodurch die Gerinnselbildung erfolgt. Hämophilie B ist eine geschlechtsgebundene, erbliche Störung der Blutgerinnung aufgrund erniedrigter Faktor-IX-Spiegel. Dies führt, entweder spontan oder in Folge unfallbedingter oder chirurgischer Traumata zu starken Blutungen in Gelenken, Muskeln oder inneren Organen. Durch die Substitutionstherapie werden die Faktor-IX-Plasmaspiegel erhöht, wodurch eine vorübergehende Korrektur des Faktor-IX-Mangels und der Blutungsneigung erfolgt.
Kinder und Jugendliche
Die Anwendung von IMMUNINE bei Kindern unter 6 Jahren kann nicht empfohlen werden, da hierzu nur unzureichende Daten vorliegen.
5.2 Pharmakokinetische Eigenschaften
Gemäß den Ergebnissen einer Studie der Phase IV betrug die mittlere inkrementelle Recovery (IR) von Faktor IX bei vorbehandelte Patienten (PTPs) ab 12 Jahren (n = 27) 1,1 (± 0,27) I.E./dl pro
I.E./kg, mit einem Streubereich von 0,6 bis 1,7 I.E./dl pro I.E./kg. In der gleichen Studie ergab sich bei
PTPs bis einschließlich 11 Jahren (n = 4) eine mittlere IR von 0,9 (± 0,12) mit einem Streubereich von 0,8 bis 1,1.
Eine pharmakokinetische Studie mit 26 Patienten zeigte die unten beschriebenen Ergebnisse:
|
Parameter | ||||
|
Anzahl |
Mittelwert |
SD |
95 % Cl | |
|
Clearance (ml/h/kg) |
26 |
8,89 |
2,91 |
7,72 - 10,06 |
|
Mittlere Verweilzeit (h) |
26 |
23,86 |
5,09 |
1,85 - 25,88 |
Die biologische Halbwertszeit beträgt ca. 17 Stunden.
5.3 Präklinische Daten zur Sicherheit
IMMUNINE ist ein hochgereinigtes Faktor-IX-Konzentrat, das nur Spuren von Faktor II, VII und X enthält. Die einmalige Verabreichung von IMMUNINE an Versuchstieren zeigte keine Anzeichen eines toxischen oder thrombogenen Potentials.
Die Durchführung von Studien zur Toxizität bei wiederholter Gabe ist aufgrund des Verhaltens heterologer, menschlicher Proteine im Tier nicht aussagekräftig.
Da Faktor IX ein Protein menschlichen Ursprungs ist, das unter physiologischen Bedingungen im Blutkreislauf zirkuliert, sind keine reproduktionstoxischen, mutagenen oder kanzerogenen Wirkungen zu erwarten.
6. PHARMAZEUTISCHE ANGABEN
6.1 Liste der sonstigen Bestandteile
Pulver: Natriumchlorid
Natriumzitrat
Lösungsmittel: Sterilisiertes Wasser für Injektionszwecke
6.2 Inkompatibilitäten
Da keine Kompatibilitätsstudien durchgeführt wurden, darf dieses Arzneimittel, außer mit den unter Abschnitt 6.6 aufgeführten Produkten, nicht mit anderen Arzneimitteln gemischt werden.
Es dürfen ausschließlich die mitgelieferten Injektions-/Infusionssets benutzt werden, da die Therapie als Folge einer Adsorption von humanem Blutgerinnungsfaktor IX an der inneren Oberfläche mancher Injektions-/Infusionsbestecke versagen kann.
6.3 Dauer der Haltbarkeit
2 Jahre
Die chemische und physikalische Stabilität der gebrauchsfertigen Lösung von IMMUNINE wurde für
3 Stunden bei maximal 25 °C nachgewiesen. Aus mikrobiologischer Sicht sollte die Lösung dennoch unmittelbar verwendet werden, außer die Herstellung der gebrauchsfertigen Lösung schließt das Risiko einer mikrobiologischen Kontamination aus (validierte aseptische Umgebung). Wird die gebrauchsfertige Lösung nicht unverzüglich verwendet, liegen Lagerbedingungen und -zeit in der Verantwortung des Anwenders. Die gebrauchsfertige Lösung darf nicht wieder in den Kühlschrank zurückgestellt werden.
6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Im Kühlschrank lagern (2 °C bis 8 °C). Nicht einfrieren.
In der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen.
IMMUNINE kann innerhalb der Laufzeit einmalig bis zu 3 Monate bei maximal 25 °C gelagert werden. Der Beginn der Lagerung bei maximal 25 °C ist auf der Produktschachtel einzutragen. Nach Lagerung bei maximal 25 °C darf IMMUNINE nicht mehr in den Kühlschrank zurückgegeben werden, sondern ist unmittelbar zu verbrauchen oder zu verwerfen.
Lagerungsbedingungen des rekonstituierten Arzneimittels siehe Abschnitt 6.3.
6.5 Art und Inhalt des Behältnisses
Das IMMUNINE-Pulver befindet sich in einer Einzeldosis-Durchstechflasche aus Glas der hydrolytischen Klasse II, das Lösungsmittel in Einzeldosis-Durchstechflaschen aus Glas der hydrolytischen Klasse I. Die Durchstechflasche mit dem Blutgerinnungspräparat ist mit einem Chlorbutyl-, die Lösungsmittel-Durchstechflasche mit einem Brombutylgummistopfen verschlossen.
Packungsinhalt:
1 Durchstechflasche IMMUNINE 600 I.E. oder IMMUNINE 1200 I.E.
1 Durchstechflasche mit 5 ml oder 10 ml Sterilisiertem Wasser für Injektionszwecke 1 Transfernadel 1 Belüftungsnadel 1 Filternadel 1 Einmalnadel
1 Einmalspritze (5 ml oder 10 ml)
1 Infusionsset
Packungsgröße: 1 x 600 I.E. oder 1 x 1200 I.E.
6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung
Nur das beigepackte Injektions-/Infusionsset verwenden.
IMMUNINE soll erst unmittelbar vor der Verabreichung aufgelöst werden. Danach soll die Lösung unverzüglich verwendet werden (die Lösung enthält keine Konservierungsmittel). Die gebrauchsfertige Lösung ist vor der Verabreichung visuell auf Partikel oder Verfärbungen zu überprüfen. Die Lösung muss klar oder leicht opaleszent sein. Trübe Lösungen, oder solche mit Niederschlag, sind zu verwerfen.
Es wird empfohlen, einen gemeinsamen venösen Zugang vor und nach der Verabreichung von IMMUNINE mit isotonischer Kochsalz-Lösung zu spülen.
Auflösung des Pulvers zur Herstellung einer Injektionslösung:
Auf aseptische Arbeitsweise achten!
1. Erwärmen Sie die ungeöffnete Lösungsmittel-Durchstechflasche (Sterilisiertes Wasser für Injektionszwecke) auf Raumtemperatur (max. 37 °C).
2. Entfernen Sie die Schutzkappen von den Durchstechflaschen mit Pulver und Lösungsmittel (Abb. A) und desinfizieren Sie die Gummistopfen beider Durchstechflaschen.
3. Entfernen Sie durch Drehen das eine Ende der Schutzkappe von der beigepackten Transfernadel. Stechen Sie nun mit der Nadel durch den Gummistopfen der Lösungsmittel-Durchstechflasche (Abb. B und C).
4. Entfernen Sie das andere Ende der Schutzkappe von der Transfernadel. Achten Sie darauf, das freie Ende nicht zu berühren!
5. Drehen Sie die Lösungsmittel-Durchstechflasche kopfüber um und stechen Sie nun das andere Ende der Transfernadel durch den Gummistopfen der Pulver-Durchstechflasche (Abb. D). Durch das Vakuum in der Pulver-Durchstechflasche wird das Lösungsmittel angesaugt.
6. Nachdem das Lösungsmittel vollständig in die Pulver-Durchstechflasche geflossen ist, entfernen Sie die Lösungsmittel-Durchstechflasche mit der Transfernadel (Abb. E). Um den Lösungsvorgang zu beschleunigen sollte die Pulver-Durchstechflasche sanft geschwenkt werden.
7. Nachdem das Pulver vollständig gelöst ist, stechen Sie die beigepackte Belüftungsnadel ein (Abb. F), eventuell entstandener Schaum wird zusammenfallen. Danach entfernen Sie die Belüftungsnadel.
Injektion/Infusion:
Auf aseptische Arbeitsweise achten!
1. Ein Ende der Schutzkappe der beigepackten Filternadel durch Drehen und Abziehen öffnen, entfernen und die Nadel auf die sterile Einmalspritze stecken. Die Lösung in die Spritze aufziehen (Abb. G).
2. Die Filternadel von der Spritze abziehen und die Lösung langsam mit dem beigepackten Infusionsset (bzw. Einmalnadel) intravenös verabreichen (maximale Infusionsrate: 2 ml pro Minute).
Bei Infusion muss ein Einmalinfusions-Set mit einem geeigneten Filter verwendet werden.
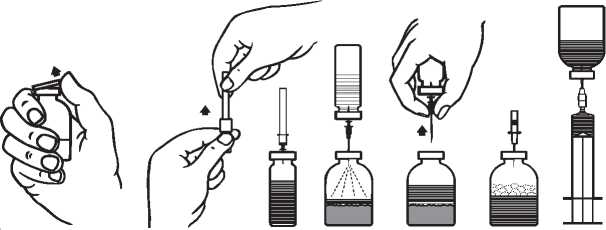
Abb. A Abb. B Abb. C Abb. D Abb. E Abb. F Abb. G
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu entsorgen.
7. INHABER DER ZULASSUNG
Baxter Deutschland GmbH Edisonstraße 4 85716 Unterschleißheim Telefon: 089/31701-0 Fax: 089/31701-177 E-Mai: info de@baxter.com
8. ZULASSUNGSNUMMER
PEI.H.03595.02.1
PEI.H.03595.03.1
9. DATUM DER ERTEILUNG DER ZULASSUNG
Datum der Erteilung der Zulassung: 05. Mai 2008 Datum der Verlängerung der Zulassung: 09. Juni 2011
10. STAND DER INFORMATION
Juni 2014
11. SONSTIGE HINWEISE
Herkunftsländer der zur Produktion verwendeten Plasmen: Deutschland, Finnland, Norwegen, Österreich, Schweden, Schweiz, Tschechien und Vereinigte Staaten von Amerika
12. VERKAUFSABGRENZUNG
Verschreibungspflichtig
Seite 11 von 11
nach dem Versuch einer Immuntoleranzinduktion