Immuseven
FACHINFORMATION
1. BEZEICHNUNG DES ARZNEIMITTELS Immuseven
600 I.E. Blutgerinnungsfaktor VII
Pulver und Lösungsmittel zur Herstellung einer Injektionslösung
2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
Immuseven ist als lyophilisiertes Pulver erhältlich, das pro Fläschchen nominal 600 I.E.1 Gerinnungsfaktor VII aus Humanplasma enthält.
Das Produkt enthält ungefähr 60 I.E./ml (600 I.E./10 ml) Gerinnungsfaktor VII aus Humanplasma, wenn es mit 10 ml sterilisiertem Wasser für Injektionszwecke aufgelöst wird.
Die Aktivität (I.E.) wird mit Hilfe des chromogenen Tests der Europäischen Pharmakopoe gegen den Internationalen Standard für Faktor VII-Konzentrate der Weltgesundheitsorganisation (WHO) bestimmt. Die spezifische Aktivität von Immuseven beträgt mindestens 2 I.E. Faktor VII/mg Protein.
Das rekonstituierte Produkt enthält weniger als 0,20 I.E. Faktor II/I.E. Faktor VII,
0,15 I.E. Faktor IX/I.E. Faktor VII und 0,35 I.E. Faktor X/I.E. Faktor VII.
Darüber hinaus ist Heparin-Natrium (maximal 0,5 I.E./I.E. Faktor VII) enthalten.
Vollständige Auflistung der sonstigen Bestandteile siehe Abschnitt 6.1.
3. DARREICHUNGSFORM
Pulver und Lösungsmittel zur Herstellung einer Injektionslösung
Weißes oder leicht gefärbtes Pulver oder bröckelige Substanz. Nach der Rekonstitution weist die Lösung einen pH-Wert zwischen 6,5 - 7,5 und eine Osmolalität von mindestens 240 mOsm/kg auf.
4. KLINISCHE ANGABEN
4.1 Anwendungsgebiete
• Behandlung von Blutungsstörungen, die durch einen isolierten angeborenen Faktor VII-Mangel verursacht werden.
• Prophylaxe von Blutungsstörungen, die durch einen isolierten, angeborenen Faktor VII-Mangel verursacht werden und die mit Blutungsereignissen, sowie einer residualen Faktor VII-C-Aktivität von weniger als 25 % des Normalwertes (0,25 I.E./ml) assoziiert sind.
Das Präparat enthält keine nennenswerte Menge an aktiviertem Faktor Vlla und sollte nicht bei Hämophilie-Patienten mit Inhibitoren verwendet werden.
4.2 Dosierung und Art der Anwendung Dosierung
Die Behandlung sollte unter Aufsicht eines in der Substitutionstherapie mit Gerinnungsfaktoren erfahrenen Arztes eingeleitet werden.
Aufgrund der Seltenheit der Erkrankung stehen nur begrenzte Daten über den klinischen Einsatz von Faktor VII-Produkten zur Verfügung. Deshalb können nur allgemeine Dosierungsrichtlinien gegeben werden, während der individuelle Dosisbedarf nur auf Basis von regelmäßigen Faktor VII-Bestimmungen und kontinuierlicher, klinischer Überwachung des Patienten festgelegt werden kann.
Dosierung und Dauer der Substitutionstherapie hängen von der Schwere des Faktor VII-Mangels sowie dem Ort und Ausmaß der Blutung und dem klinischen Zustand des Patienten ab. Der Zusammenhang zwischen dem individuell noch vorhandenen Faktor VII-Spiegel und der klinischen Blutungsneigung ist geringer als bei der klassischen Hämophilie.
Die Anzahl der verabreichten Faktor VII-Einheiten ist in Internationalen Einheiten (I.E.) angegeben, die sich auf den aktuellen Standard der WHO für Faktor VII-Produkte beziehen. Die Faktor VII-Aktivität im Plasma wird entweder prozentual (relativ zu normalem Humanplasma) oder in Internationalen Einheiten (bezogen auf den Internationalen Standard für Faktor VII-Konzentrate) ausgedrückt.
Eine Internationale Einheit (I.E.) Faktor VII-Aktivität entspricht der Aktivität an Faktor VII in 1 ml normalem Humanplasma. Die Berechnung der erforderlichen Dosis Faktor VII beruht auf der Erkenntnis, dass 1 Internationale Einheit (I.E.) Faktor VII pro kg Körpergewicht die Faktor VII-Aktivität im Plasma um ca. 1,9 % (1,9 I.E./dl) der normalen Aktivität erhöht.
Die erforderliche Dosis wird mit folgender Formel ermittelt:
Erforderliche Einheiten = Körpergewicht (kg) x gewünschter Faktor VII-Anstieg (% oder I.E./ml) x 0,5*_
Wenn die individuelle Recovery bekannt ist, sollte dieser Wert anstelle des Wertes 0,5 für die Berechnung eingesetzt werden.
Die Dosis und das Dosierungsintervall sollten sich immer nach der klinischen Wirksamkeit im Einzelfall richten. Dies ist besonders wichtig bei der Behandlung des Faktor VII-Mangels, da dort die individuelle Blutungsneigung nicht streng mit der im Plasma durch Labortests ermittelten Faktor VII-Aktivität korreliert. Die Dosierungsintervalle müssen an die kurze Zirkulationshalbwertszeit von Faktor VII von ungefähr 3 bis 5 Stunden angepasst werden. Wenn Immuseven durch intermittierende Injektionen/Infusionen verabreicht wird, sind oft Dosierungsintervalle von 6 - 8 Stunden ausreichend. Normalerweise sind bei der Behandlung des Faktor VII-Mangels niedrigere Werte des Mangelfaktors in Bezug auf die Aktivität im Normalplasma erforderlich als bei der klassischen Hämophilie (Hämophilie A und B). Die folgende Tabelle gibt einige Hinweise zur Verabreichung intermittierender Injektionen/Infusionen auf Basis von begrenzten, klinischen Erfahrungen. Es existieren keine medizinischen Erkenntnisse aus klinischen Wirksamkeitsstudien.
|
Grad der Blutung/ Art des chirurgischen Eingriffs |
Angestrebte Faktor VII-Aktivität [I.E./ml]* |
Dosierungsfrequenz [Stunden] Dauer der Therapie [Tagen] |
|
Kleinere Blutungen |
0,10 - 0,20 |
eine Einzeldosis |
|
Schwere Blutung |
0,25 - 0,40 (Plasmaspiegel) |
für 8 bis 10 Tage oder bis zur vollständigen Heilung** |
|
Kleinere chirurgische Eingriffe |
0,20 - 0,30 |
eine Einzeldosis vor dem Eingriff oder, wenn das Blutungsrisiko höher eingeschätzt wird, bis zur Wundheilung |
|
Größere chirurgische Eingriffe |
präoperativ >0,50 dann 0,25 - 0,45 (Plasmaspiegel) |
für 8 - 10 Tage oder bis zur kompletten Wundheilung** |
* I.E./ml = 100 I.E./dl = 100 % des Normalplasmas. Die Faktor VII-Aktivität im Plasma wird entweder als Prozentsatz (bezogen auf Normalplasma) oder in Internationalen Einheiten (bezogen auf den Internationalen Standard für Faktor VII-Konzentrate im Plasma) angegeben.
** Basierend auf der klinischen Einschätzung können in Einzelfällen gegen Ende der Behandlung niedrigere Dosen ausreichend sein, vorausgesetzt dass eine adäquate Blutstillung erreicht wird.
Wenn hohe Faktor VII-Spiegel über eine längere Zeit aufrechterhalten werden sollen, ist alle 8 - 12 Stunden eine Dosis erforderlich.
Kinder und Jugendliche
Die Anwendung von Immuseven bei Kindern unter 6 Jahren kann nicht empfohlen werden, da hierzu nur unzureichende Daten vorliegen.
Art der Anwendung
Die Rekonstitution des Arzneimittels erfolgt wie in Abschnitt 6.6 beschrieben. Die gebrauchsfertige Lösung wird langsam intravenös verabreicht. Eine Infusionsgeschwindigkeit von 2 ml pro Minute sollte nicht überschritten werden.
4.3 Gegenanzeigen
• Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile
• Hohes Risiko für eine Thrombose oder disseminierte intravasale Gerinnung (DIC) (siehe Abschnitt 4.4)
• Bekannte Allergie gegen Heparin oder Fälle von Heparin-induzierter Thrombozytopenie in der Vorgeschichte
4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Warnhinweise
Im Zusammenhang mit der Anwendung Faktor VII-haltiger Produkte wurde über Überempfindlichkeitsreaktionen, einschließlich anaphylaktische Reaktionen, berichtet.
Überempfindlichkeitsreaktionen wurden auch bei Immuseven beobachtet (siehe Abschnitt 4.8).
Wenn allergische oder anaphylaktische Reaktionen auftreten, muss die Verabreichung sofort abgebrochen werden. Im Falle eines Schocks ist eine Schockbehandlung nach aktuellem medizinischem Standard durchzuführen. Patienten und/oder ihre Begleitpersonen müssen über frühe Anzeichen von Überempfindlichkeitsreaktionen aufgeklärt werden. Wenn diese Symptome auftreten, müssen die Patienten angewiesen werden, die Behandlung sofort abzubrechen und ihren Arzt zu kontaktieren.
Als Plasmaderivat enthält das Faktor VII-Konzentrat auch andere Proteine menschlichen Ursprungs.
Standardmaßnahmen zur Vorbeugung von Infektionen, die sich durch den Einsatz von Arzneimitteln ergeben, die aus Blut oder Blutplasma hergestellt sind, schließen die Auswahl der Spender und das Screening der einzelnen Blutspenden und Plasmapools auf spezifische Infektionsmarker sowie effektive Schritte zur Inaktivierung/Eliminierung von Viren im Herstellungsverfahren ein. Dennoch kann bei der Verabreichung von Arzneimitteln aus menschlichem Blut oder Blutplasma die Möglichkeit der Übertragung von Krankheitserregern nicht völlig ausgeschlossen werden. Dies gilt auch für bislang unbekannte oder neu aufgetretene Viren und andere Pathogene.
Die getroffenen Maßnahmen werden als wirksam erachtet für umhüllte Viren wie das Immunschwächevirus (HIV), das Hepatitis B-Virus (HBV) und Hepatitis C-Virus (HCV), und für das nicht-umhüllte Hepatitis A-Virus (HAV). Für nicht-umhüllte Viren wie Parvovirus B19 können diese Maßnahmen möglicherweise nur begrenzt wirksam sein. Parvovirus B19-Infektionen können schwerwiegende Folgen für Schwangere (Infektion des Fötus) und für Menschen mit Immunmangelkrankheiten oder gesteigerter Erythropoese (z. B. hämolytische Anämie) haben (siehe Abschnitt 4.6).
Für Patienten, die regelmäßig aus Blutplasma hergestellte Faktor VII-Präparate erhalten, wird grundsätzlich eine Impfung gegen Hepatitis A und B empfohlen.
Es wird dringend empfohlen, bei jeder Verabreichung von Immuseven, die Bezeichnung (Immuseven) und die Chargennummer des Produktes zu notieren, damit eine Verbindung zwischen dem Patienten und der angewendeten Charge hergestellt werden kann.
Bei der Behandlung mit einem Gerinnungsfaktor VII-haltigen Produkt aus Humanplasma besteht das Risiko einer Thrombose oder einer disseminierten intravasalen Gerinnung (DIC). Im Zusammenhang mit Immuseven wurden zerebrale Venenthrombosen, tiefe Venenthrombosen und Thrombophlebitis beobachtet. Patienten, die mit Gerinnungsfaktor VII behandelt werden, müssen daher sorgfältig auf Anzeichen oder Symptome einer intravasalen Gerinnung oder Thrombose hin überwacht werden. Wegen des Risikos von thromboembolischen Komplikationen sollten höhere Dosen von Gerinnungsfaktor VII-Konzentraten aus Humanplasma bei
• Patienten mit einer Vorgeschichte einer koronaren Herzerkrankung
• Patienten mit einer Lebererkrankung
• postoperativen Patienten
• Neugeborenen
• Risikopatienten für thromboembolische Ereignisse oder DIC
nur mit Vorsicht verabreicht werden. In all diesen Fällen sollte der Nutzen der Behandlung mit Immuseven gegen das Risiko möglicher Komplikationen abgewogen werden.
Die Substitutionstherapie mit humanem Faktor VII - einschließlich Immuseven - kann zur Bildung zirkulierender Antiköper gegen Faktor VII führen. Falls solche Inhibitoren auftreten, zeigt sich dies in einer unzureichenden klinischen Wirksamkeit.
Vorsichtsmaßnahmen
Immuseven enthält 1,56 bis 1,90 mmol (ca. 40 mg) Natrium pro Durchstechflasche. Dies ist bei Patienten zu berücksichtigen, die auf eine natriumarme Ernährung achten müssen.
Es wird auf die Dokumentationspflicht gemäß Transfusionsgesetz hingewiesen.
4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Es sind keine Wechselwirkungen zwischen Gerinnungsfaktor VII-Produkten aus Humanplasma und anderen Arzneimitteln bekannt.
Auswirkungen auf serologische Untersuchungen:
Bei der Durchführung von Gerinnungstests, die bei Patienten mit hohen Dosierungen von Immuseven auf Heparin ansprechen, muss die mit dem Produkt verabreichte Heparin-Konzentration berücksichtigt werden.
4.6 Fertilität, Schwangerschaft und Stillzeit
Die Auswirkungen von Immuseven auf die Fertilität wurden nicht in kontrollierten klinischen Studien untersucht.
Die Sicherheit von Immuseven bei der Anwendung während einer Schwangerschaft wurde nicht in kontrollierten klinischen Studien nachgewiesen.
Tierversuche eignen sich nicht dazu, die Sicherheit bezüglich Schwangerschaftsverlauf, embryonaler und fetaler Entwicklung, Entbindung oder postnataler Entwicklung zu beurteilen. Daher sollte ein Faktor VII-Konzentrat einer schwangeren Frau nur bei strenger Indikationsstellung und nach Abwägung der potenziellen Risiken verschrieben werden.
Zum Risiko einer Infektion mit Parvovirus B19 während der Schwangerschaft siehe Abschnitt 4.4.
4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Es sind keine Auswirkungen auf die Verkehrstüchtigkeit und das Bedienen von Maschinen bekannt.
4.8 Nebenwirkungen
Die folgenden Nebenwirkungen beruhen auf Berichten aus klinischen Studien und auf der Marktbeobachtung von Immuseven:
Die Häufigkeit der Nebenwirkungen wurde nach den folgenden Kriterien angegeben: Sehr häufig (>1/10), häufig (>1/100; <1/10), gelegentlich (>1/1.000; <1/100), selten
(>1/10.000; <1/1.000), sehr selten (<1/10.000), nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
Die folgenden Nebenwirkungen wurden in einer klinischen Studie mit 57 Kindern und Erwachsenen mit angeborenem Faktor VII-Mangel beobachtet, die Faktor VII-Konzentrate zur Behandlung von akuten Blutungen, bei chirurgischen Eingriffen und zur Langzeitprophylaxe von Blutungsereignissen erhielten. Im Laufe dieser Studie wurden 8234 Expositionstage erreicht.
In der nachfolgenden Tabelle sind die beobachteten Nebenwirkungen aus der klinischen Prüfung und der Post-Marketing-Überwachung nach MedDRA-Systemorganklassen (SOC) aufgelistet und, soweit möglich, mit der bevorzugten Bezeichnung nach Schweregrad geordnet.
|
Systemorganklassen gemäß MedDRA-Datenbank |
Nebenwirkung |
Häufigkeit |
|
Erkrankungen des Blutes und des Lymphsystems |
Positiver Nachweis von antiFaktor VN-Antikörpern |
Nicht bekannt |
|
Erkrankungen des Immunsystems |
Überempfindlichkeitsreaktion |
Nicht bekannt |
|
Psychiatrische Erkrankungen |
Verwirrtheit Schlaflosigkeit Unruhe |
Nicht bekannt |
|
Erkrankungen des Nervensystems |
Zerebrale Venenthrombose Schwindel Dysästhesie Kopfschmerzen |
Nicht bekannt |
|
Herzerkrankungen |
Herzrhythmusstörungen Hypotonie |
Nicht bekannt |
|
Gefäßerkrankungen |
Hautrötung Tiefe Venenthrombosen Oberflächliche Thrombophlebitis Hitzewallungen |
Häufig1 Nicht bekannt Nicht bekannt Nicht bekannt |
|
Erkrankungen der Atemwege, des Brustraums und Mediastinums |
Bronchospasmus Dyspnoe |
Nicht bekannt |
|
Erkrankungen des Gastrointestinaltrakts |
Durchfall Übelkeit |
Nicht bekannt |
|
Erkrankungen der Haut und des Unterhautzellgewebes |
Ausschlag Juckreiz |
Häufig1 Nicht bekannt |
|
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort |
Fieber Brustschmerzen Anomales Gefühl2 Engegefühl in der Brust |
Häufig1 Häufig1 Häufig1 Nicht bekannt |
1
Die Häufigkeit beruht auf der Anzahl der Patienten, bei denen das Auftreten dieser Nebenwirkung mindestens in einem möglichen Zusammenhang betrachtet wurde.
2 Beinhaltet auch Benommenheitszustände.
Zur Sicherheit vor übertragbaren Erregern siehe Abschnitt 4.4. Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-
Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel, Paul-Ehrlich-Institut, Paul-Ehrlich-Str. 51-59, 63225 Langen, Tel: +49 6103 77 0, Fax: +49 6103 77 1234, Website: www.pei.de anzuzeigen.
4.9 Überdosierung
Die Gabe von hohen Dosen an Faktor VII-haltigen Produkten (ProthrombinkomplexProdukten) wird mit Fällen von Myokardinfarkten, disseminierter intravasaler Gerinnung, Venenthrombosen und Lungenembolien in Verbindung gebracht.
Daher steigt im Fall einer Überdosierung das Risiko für das Auftreten thromboembolischer Komplikationen.
5. PHARMAKOLOGISCHE EIGENSCHAFTEN
5.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Antihämorrhagika: Blutgerinnungsfaktor VII ATC-Code: B02BD05
Faktor VII gehört zu den Vitamin-K-abhängigen Gerinnungsfaktoren im normalen Humanplasma. Es ist ein Einzelketten-Glycoprotein mit einem Molekulargewicht von etwa 50.000 Dalton. Faktor VII ist das Zymogen der aktiven Serinprotease Faktor VIIa, durch die der extrinsische Weg der Reaktionskette der Blutgerinnung ausgelöst wird. Der Komplex aus Gewebefaktor und Faktor VIIa aktiviert die Blutgerinnungsfaktoren IX und X, wodurch die Faktoren IXa und Xa gebildet werden. Mit dem weiteren Fortschreiten der Gerinnungskaskade wird schließlich Thrombin gebildet, Fibrinogen wird in Fibrin umgewandelt und ein Gerinnsel entsteht. Die normale Bildung von Thrombin ist auch für die Plättchenfunktion als Teil der primären Hämostase von vitaler Bedeutung. Ein angeborener Faktor VII-Mangel ist eine autosomal-rezessive Erbkrankheit. Die Gabe von humanem Faktor VII erhöht die Plasmakonzentration an Faktor VII und kann so zeitweise den Gerinnungsdefekt bei Patienten mit Faktor VII-Mangel beheben.
5.2 Pharmakokinetische Eigenschaften
Nach der intravenösen Gabe von Immuseven liegt die erwartete in-vivo-Recovery bei ungefähr 60 bis 100 %, mit einer mittleren Halbwertszeit von ca. 3 bis 5 Stunden.
Die folgende Tabelle fasst die Ergebnisse einer pharmakokinetischen Studie über „Recovery und Halbwertszeit von dampfbehandeltem Faktor VII-Konzentrat"2 zusammen. Sie enthält pharmakokinetische Daten über die zunehmende Recovery (IR), die Fläche unter der Kurve (AUC), die mittlere Verweildauer (MRT), die Clearance (Cl), das Verteilungsvolumen im Gleichgewichtszustand (Vss) und über die Halbwertszeit (HL) sowohl in der Anfangsphase (HL1) als auch in der Eliminationsphase (HL2).
Ergebnisse:
|
IR |
AUC |
MRT |
Cl |
Vss |
HL |
HL1 |
HL2 | |
|
Min |
1,6 |
1,9 |
3,8 |
100 |
503 |
2,7 |
0,21 |
2,5 |
|
Q1 |
1,7 |
3,9 |
5,5 |
206 |
1345 |
3,8 |
0,68 |
2,7 |
|
Median |
1,9 |
4,3 |
6,9 |
326 |
1893 |
4,8 |
1,19 |
3,1 |
|
Q3 |
3,0 |
7,2 |
7,4 |
396 |
3377 |
5,1 |
1,87 |
5,3 |
|
Max |
3,4 |
9,8 |
15,1 |
531 |
6410 |
10,5 |
2,79 |
10,8 |
5.3 Präklinische Daten zur Sicherheit
Der in Immuseven enthaltene humane Blutgerinnungsfaktor VII ist ein normaler Bestandteil des menschlichen Plasmas und verhält sich wie körpereigener Faktor VII.
Die Prüfung auf Toxizität nach einmaliger Verabreichung ist nicht durchzuführen, da höhere Dosen zu einer Volumenüberlastung führen. Toxizitätsuntersuchungen bei wiederholter Verabreichung an Tieren sind wegen einer immunologischen Reaktion auf heterologe Proteine und der Beeinträchtigung von Folgetests ungeeignet.
Da klinische Erfahrungen keinen Hinweis auf kanzerogene und mutagene Wirkungen geben, werden experimentelle Studien, insbesondere bei heterologen Spezies, nicht als notwendig erachtet.
6. PHARMAZEUTISCHE ANGABEN
6.1 Liste der sonstigen Bestandteile
Pulver:
Natriumcitrat
Natriumchlorid
Heparin-Natrium
Lösungsmittel:
Wasser für Injektionszwecke
6.2 Inkompatibilitäten
Dieses Arzneimittel darf nicht mit anderen Arzneimitteln oder Heparin gemischt werden.
Es darf nur das mitgelieferte Injektionsset verwendet werden, da es bei Verwendung anderer Verabreichungssets durch Adsorption des Blutgerinnungsfaktors VII an den inneren Oberflächen zu einem Behandlungsfehler kommen kann.
6.3 Dauer der Haltbarkeit
3 Jahre
Die chemische und physikalische Stabilität der rekonstituierten Lösung ist bei Raumtemperatur (nicht über 25 °C) über 3 Stunden belegt. Aus mikrobiologischer Sicht sollte Immuseven jedoch unmittelbar nach der Rekonstitution verabreicht werden. Wird die gebrauchsfertige Lösung nicht sofort verwendet, ist der Anwender für die Lagerungsbedingungen und -dauer verantwortlich. Die gebrauchsfertige Lösung darf nicht mehr gekühlt werden.
6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Im Kühlschrank lagern (2 °C bis 8 °C). Nicht einfrieren.
In der Originalpackung aufbewahren, um das Produkt vor Licht zu schützen.
Immuseven darf innerhalb der angegebenen Laufzeit einmal bis zu maximal 6 Monaten bei Raumtemperatur (nicht über 25 °C) aufbewahrt werden. Der Beginn der Raumtemperaturlagerung sollte auf dem Umkarton vermerkt werden. Nach der Lagerung bei Raumtemperatur muss Immuseven sofort verbraucht oder verworfen werden. Es darf nicht wieder gekühlt werden.
Aufbewahrungsbedingungen nach Rekonstitution des Arzneimittels, siehe Abschnitt 6.3.
6.5 Art und Inhalt des Behältnisses
600 I.E. Blutgerinnungsfaktor VII als gefriergetrocknetes Pulver in einer Durchstechflasche (Glas Typ II) mit Gummi-Stopfen .
10 ml Wasser für Injektionszwecke in einer Durchstechflasche (Glas Typ I) mit GummiStopfen.
Jede Packung enthält außerdem ein Set zum Auflösen und Verabreichen, bestehend aus 1 Einmalspritze, 1 Einmalnadel, 1 Transfernadel, 1 Filternadel, 1 Belüftungsnadel und 1 Flügelkanüle.
6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung
Immuseven erst unmittelbar vor der Verabreichung auflösen. Nur das mitgelieferte Injektionsset verwenden.
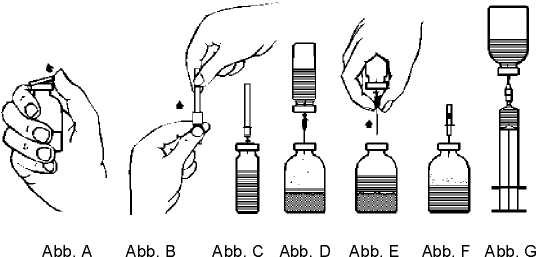
Auflösen der Trockensubstanz
1. Das ungeöffnete Lösungsmittel-Fläschchen auf Raumtemperatur bringen (max. 37 °C).
2. Die Schutzkappen von den Flaschen mit Pulver und Lösungsmittel (Abb. A) entfernen und die Gummistopfen beider Flaschen reinigen.
3. Die Schutzkappe von einem Ende der mitgelieferten Transfernadel durch Drehen und Ziehen entfernen (Abb. B). Die freigelegte Nadel durch den Gummistopfen der Lösungsmittelflasche stechen (Abb. C).
4. Die Schutzkappe von der anderen Seite der Transfernadel abziehen, ohne das freie Ende zu berühren.
5. Die Lösungsmittelflasche kopfüber über die Konzentratflasche halten und das freie Ende der Transfernadel durch den Gummistopfen in der Konzentratflasche stechen (Abb. D). Durch das entstehende Vakuum wird das Lösungsmittel angesaugt.
6. Die beiden Flaschen trennen, indem Sie die Transfernadel von der Konzentratflasche entfernen (Abb. E). Den Lösungsvorgang durch sanftes und gleichmäßiges Schwenken der Konzentratflasche beschleunigen.
7. Ist das Pulver vollständig aufgelöst, die mitgelieferte Belüftungsnadel (Abb. F) einführen, so dass eventuell vorhandener Schaum zusammenfällt. Anschließend die Belüftungsnadel wieder entfernen.
Injektion
1. Die Schutzkappe der beiliegenden Filternadel durch Drehen und Ziehen entfernen und die Nadel auf eine Einmalspritze setzen. Die Lösung in die Spritze aufziehen (Abb. G).
2. Die Filternadel von der Spritze trennen und die Lösung mit Hilfe der mitgelieferten Flügelkanüle (oder der mitgelieferten Einmalnadel) langsam intravenös applizieren (maximale Injektionsrate: 2 ml/Min).
3. Im Fall einer häuslichen Behandlung und Selbstverabreichung, stellen Sie sicher, dass gebrauchte Nadeln und Spritzen in den Karton des Auflösungssets zurückgelegt werden und geben Sie diesen Karton an Ihr Hämophilie-Zentrum zurück.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Die Lösung muss farblos oder schwach-gelb bis bräunlich sein. Trübe Lösungen oder solche mit Niederschlag sollten nicht verwendet werden. Die rekonstituierte Lösung soll vor Verabreichung visuell auf Schwebeteilchen oder Verfärbung überprüft werden.
Immuseven sollte nach der Rekonstitution sofort verabreicht werden.
7. INHABER DER ZULASSUNG
Baxter Deutschland GmbH Edisonstraße 4
85716 Unterschleißheim Telefon: 089/31701-0 Fax: 089/31701-177 E-Mail: info_de@baxter.com
8. ZULASSUNGSNUMMER
PEI.H.03341.01.1
9. DATUM DER ERTEILUNG DER ZULASSUNG/VERLÄNGERUNG DER ZULASSUNG
22. November 2005
10. STAND DER INFORMATION
August 2013
11. SONSTIGE INFORMATIONEN
Herkunftsländer der zur Produktion verwendeten Plasmen
Deutschland, Finnland, Norwegen, Österreich, Schweden, Schweiz, Tschechien und Vereinigte Staaten von Amerika.
12. VERKAUFSABGRENZUNG
Verschreibungspflichtig
Baxter ist eine Marke der Baxter International Inc.
Seite 11/11
reziproke, beobachtete Recovery (ml/kg)
Rivard GE et al 1994. Clinical study of recovery and half-life of vapor-heated factor VII concentrate. Transfusion 1994; 34:975-979
Engl, W.: Statistical Report for the Clinical Study of Recovery and Half-Life of factor VII Concentrate (Human) IMMUNO,
Vapor Heated dated August 25, 2006