Lodotra 5 Mg Tabletten Mit Veränderter Wirkstofffreisetzung
Wortlaut der für die Fachinformation vorgesehenen Angaben
F achinform ation
1. BEZEICHNUNG DER ARZNEIMITTEL
Lodotra 1 mg Tabletten mit veränderter Wirkstofffreisetzung Lodotra 2 mg Tabletten mit veränderter Wirkstofffreisetzung Lodotra 5 mg Tabletten mit veränderter Wirkstofffreisetzung
2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
Lodotra 1 mg Tabletten mit veränderter Wirkstofffreisetzung
Eine Tablette mit veränderter Wirkstofffreisetzung enthält 1 mg Prednison.
Sonstiger Bestandteil mit bekannter Wirkung: Lactose
Eine Tablette mit veränderter Wirkstofffreisetzung enthält 42,80 mg Lactose.
Lodotra 2 mg Tabletten mit veränderter Wirkstofffreisetzung
Eine Tablette mit veränderter Wirkstofffreisetzung enthält 2 mg Prednison.
Sonstiger Bestandteil mit bekannter Wirkung: Lactose
Eine Tablette mit veränderter Wirkstofffreisetzung enthält 41,80 mg Lactose.
Lodotra 5 mg Tabletten mit veränderter Wirkstofffreisetzung
Eine Tablette mit veränderter Wirkstofffreisetzung enthält 5 mg Prednison.
Sonstiger Bestandteil mit bekannter Wirkung: Lactose
Eine Tablette mit veränderter Wirkstofffreisetzung enthält 38,80 mg Lactose.
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
3. DARREICHUNGSFORM
Tablette mit veränderter Wirkstofffreisetzung
Lodotra 1 mg Tabletten mit veränderter Wirkstofffreisetzung
Blassgelblich-weiße, zylinderförmige Tablette, 5 mm hoch mit 9 mm Durchmesser, mit veränderter Wirkstofffreisetzung mit der Prägung „NP1“ auf einer Seite.
Lodotra 2 mg Tabletten mit veränderter Wirkstofffreisetzung
Gelblich-weiße, zylinderförmige Tablette, 5 mm hoch mit 9 mm Durchmesser, mit veränderter Wirkstofffreisetzung mit der Prägung „NP2“ auf einer Seite.
Lodotra 5 mg Tabletten mit veränderter Wirkstofffreisetzung
Hellgelbe, zylinderförmige Tablette, 5 mm hoch mit 9 mm Durchmesser, mit veränderter Wirkstofffreisetzung mit der Prägung „NP5“ auf einer Seite.
4.1 Anwendungsgebiete
Lodotra wird angewendet bei Erwachsenen zur Behandlung der mäßigen bis schweren, aktiven rheumatoiden Arthritis, insbesondere wenn diese von morgendlicher Gelenksteifigkeit begleitet ist.
4.2 Dosierung und Art der Anwendung
Dosierung
Die angemessene Dosierung richtet sich nach der Schwere der Erkrankung und dem individuellen Ansprechen des Patienten. Zur Einleitung der Therapie wird in der Regel eine Dosis von 10 mg Prednison empfohlen. In bestimmten Fällen kann eine höhere Anfangsdosis erforderlich sein (z. B.
15 oder 20 mg Prednison). In Abhängigkeit von der klinischen Symptomatik und dem Ansprechen des Patienten kann die Anfangsdosis schrittweise auf eine niedrigere Erhaltungsdosis reduziert werden.
Bei der Umstellung vom Standard-Behandlungsschema (morgendliche Glucocorticoidgabe) auf die abendliche Einnahme von Lodotra gegen 22 Uhr sollte die gleiche Dosis (bezogen auf mg PrednisonÄquivalent) beibehalten werden. Nach der Umstellung kann die Dosis entsprechend der klinischen Situation angepasst werden.
Für Dosierungen, die mit dieser Stärke nicht erreichbar/praktikabel sind, stehen andere Stärken dieses Arzneimittels zur Verfügung.
Für die Langzeitbehandlung der rheumatoiden Arthritis kann je nach Schwere des Krankheitsverlaufs eine Dosis von bis zu 10 mg Prednison täglich verwendet werden.
In Abhängigkeit vom Behandlungserfolg kann die Dosis alle 2-4 Wochen in Schritten von 1 mg verringert werden, bis eine angemessene Erhaltungsdosis erreicht ist.
Wenn Lodotra abgesetzt werden soll, ist die Dosis alle 2-4 Wochen um 1 mg zu reduzieren, dabei sind bei Bedarf Parameter des adrenergen Regelkreises zu überwachen.
Kinder und Jugendliche
Da bei Kindern und Jugendlichen zur Verträglichkeit und Wirksamkeit keine ausreichenden Daten vorliegen, wird die Anwendung hier nicht empfohlen.
Art der Anwendung
Lodotra ist am Abend (gegen 22 Uhr) mit oder nach dem Abendessen im Ganzen und unzerkaut mit ausreichend Flüssigkeit einzunehmen. Wenn das Abendessen mehr als 2-3 Stunden zurückliegt, wird empfohlen, Lodotra mit einer leichten Zwischenmahlzeit (z. B. einer Scheibe Brot mit Wurst oder Käse) einzunehmen. Lodotra darf nicht nüchtern eingenommen werden, da sonst die Bioverfügbarkeit von Prednison verringert sein kann.
Lodotra ist darauf ausgelegt, den Wirkstoff mit einer Verzögerung von etwa 4-6 Stunden nach der Einnahme freizusetzen. Die Freisetzung des Wirkstoffs und die pharmakologischen Wirkungen setzen während der Nacht ein.
Lodotra Tabletten mit veränderter Wirkstofffreisetzung bestehen aus einem prednisonhaltigen Kern und einem inerten Überzug. Für die verzögerte Freisetzung von Prednison muss der Tablettenüberzug unbeschädigt sein. Deshalb dürfen die Tabletten mit veränderter Wirkstofffreisetzung nicht zerbrochen, geteilt oder zerkaut werden.
Bei Patienten mit Hypothyreose oder Leberzirrhose kann bereits eine vergleichsweise niedrige Dosis ausreichend sein bzw. eine Dosissenkung erforderlich werden.
4.3 Gegenanzeigen
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Eine Pharmakotherapie mit Prednison sollte nur unter strengster Indikationsstellung durchgeführt werden und es sollte eine geeignete Begleittherapie mit Antiinfektiva bei Vorliegen folgender Umstände erfolgen:
- akute Virusinfektion (Herpes Zoster, Herpes simplex, Varizellen, Keratitis herpetica)
- HBsAg-positive chronisch-aktive Hepatitis
- ca. 8 Wochen vor und 2 Wochen nach Immunisierung mit Lebendimpfstoffen
- systemische Mykosen und Parasitosen (z. B. Nematoden)
- Poliomyelitis
- Lymphadenitis nach BCG-Impfung
- akute und chronische bakterielle Infektionen
- Tuberkulose in der Vorgeschichte (Achtung: Reaktivierung!) - Aufgrund ihrer immunsuppressiven Eigenschaften können Glucocorticoide Infektionen induzieren oder verstärken. Betroffene Patienten sind sorgfältig zu überwachen, z. B. mittels Tuberkulintest. Patienten mit besonderem Risiko sind mit Tuberkulostatika zu behandeln.
Darüber hinaus sollte Prednison nur unter strenger Indikationsstellung gegeben und falls erforderlich durch eine entsprechende Begleittherapie ergänzt werden bei Vorliegen folgender Umstände:
- Magen-Darm-Ulzera
- schwere Osteoporose und Osteomalazie
- schwer einzustellende Hypertonie
- schwerer Diabetes mellitus
- psychiatrische Erkrankungen (auch in der Vorgeschichte des Patienten)
- Eng- und Weitwinkelglaukom
- Hornhautulzera und -verletzungen.
Wegen der Gefahr einer Darmperforation darf Prednison nur bei zwingender Indikation und unter ausreichender Überwachung angewendet werden in folgenden Fällen:
- schwere Colitis ulcerosa mit drohender Perforation
- Divertikulitis
- Enteroanastomosen (unmittelbar postoperativ).
Lodotra kann den gewünschten Prednison-Blutspiegel nicht erreichen, wenn es nüchtern eingenommen wird. Deshalb sollte Lodotra immer mit oder nach dem Abendessen eingenommen werden, um eine ausreichende Wirksamkeit zu gewährleisten. Darüber hinaus können niedrige Plasmakonzentrationen auftreten, auch wenn Lodotra ordnungsgemäß eingenommen wurde. Im Durchschnitt aller PK Studien traten niedrige Plasmakonzentrationen bei 6 % - 7 % der verabreichten Lodotra-Dosen auf. In einer PK Studie wurde dies bei 11% der Lodotra-Dosen beobachtet. Dies sollte berücksichtigt werden, wenn die Wirkung von Lodotra nicht ausreichend ist. In diesem Fall sollte eine Umstellung auf eine herkömmliche Darreichungsform mit sofortiger Wirkstofffreisetzung in Erwägung gezogen werden.
Die Anwendung des Arzneimittels Lodotra kann bei Dopingkontrollen zu positiven Ergebnissen führen.
Lodotra sollte nicht durch Prednison-Tabletten mit sofortiger Wirkstofffreisetzung unter Beibehaltung des gleichen Einnahmeschemas ersetzt werden, da Lodotra eine verzögerte Wirkstofffreisetzung besitzt.
Bei Substitution, Beendigung oder Absetzen einer Langzeittherapie sind folgende Risiken zu bedenken: Reaktivierung der rheumatoiden Arthritis, akute Nebenniereninsuffizienz (insbesondere unter besonderer Belastung, z. B. während Infektionen, nach Unfällen, bei verstärkter körperlicher Anstrengung), Cortison-Entzugssyndrom.
Aufgrund seiner pharmakologischen Eigenschaften sollte Lodotra nicht bei akuten Indikationen anstelle von Prednison-Tabletten mit sofortiger Wirkstofffreisetzung angewendet werden.
Während der Anwendung von Lodotra muss ein möglicherweise erhöhter Bedarf an Insulin oder oralen Antidiabetika in Betracht gezogen werden. Patienten mit Diabetes mellitus sollten daher während der Behandlung engmaschig überwacht werden.
Bei Patienten mit schwer einstellbarem Bluthochdruck ist während der Behandlung mit Lodotra eine regelmäßige Blutdruckkontrolle erforderlich.
Patienten mit schwerer Herzinsuffizienz sind engmaschig zu überwachen, da die Gefahr einer Verschlechterung besteht.
Es ist Vorsicht angeraten, wenn Corticosteroide, einschließlich Prednison, an Patienten verschrieben wird, die kürzlich einen Herzinfarkt hatten, da das Risiko einer Myokardruptur besteht.
Es ist Vorsicht angeraten, wenn Corticosteroide, einschließlich Prednison, an Patienten verschrieben wird, die unter einer Niereninsuffizienz leiden.
Schlafstörungen treten bei Einnahme von Lodotra häufiger auf als bei herkömmlichen, am Morgen eingenommenen Darreichungsformen mit sofortiger Wirkstofffreisetzung. Falls Schlaflosigkeit auftritt und sich nicht bessert, kann ein Wechsel zu einer herkömmlichen Darreichungsform mit sofortiger Wirkstofffreisetzung ratsam sein.
Die Behandlung mit Lodotra kann die Anzeichen einer bestehenden oder sich entwickelnden Infektion maskieren und so die Diagnose erschweren.
Auch in niedrigen Dosierungen führt die Langzeitbehandlung mit Lodotra zu einem Anstieg des Infektionsrisikos. Solche Infektionen können auch von Mikroorganismen verursacht werden, die unter normalen Umständen nur selten Infektionen hervorrufen (so genannte opportunistische Infektionen).
Bestimmte Viruserkrankungen (Varizellen, Masern) können bei Patienten, die mit Glucocorticoiden behandelt werden, schwerer verlaufen. Besonders gefährdet sind immunsupprimierte Personen ohne bisherige Varizellen- oder Maserninfektion. Wenn diese Personen während einer Behandlung mit Lodotra Kontakt zu Masern- oder Windpockenkranken haben, sollte bei Bedarf eine vorbeugende Behandlung eingeleitet werden.
Bei Patienten mit Verdacht auf oder bestätigter Strongyloidiasis (Zwergfadenwurminfektion) können Glucocorticoide zu Hyperinfektion und einer umfassenden Vermehrung der Parasiten führen.
Impfungen mit inaktivierten Vakzinen sind grundsätzlich möglich. Es ist jedoch zu berücksichtigen, dass die Immunreaktion und somit der Erfolg der Impfung unter höheren Glucocorticoiddosen beeinträchtigt sein können.
Bei Langzeittherapie mit Lodotra sind regelmäßige ärztliche Kontrolluntersuchungen (einschließlich augenärztlicher Untersuchungen alle drei Monate) indiziert; bei vergleichsweise hohen Dosen ist auf eine ausreichende Kaliumzufuhr und auf Natriumrestriktion zu achten und der Serum-Kalium-Spiegel zu überwachen.
Kommt es während der Behandlung mit Lodotra durch besondere Ereignisse (Unfälle, Operationen etc.) zu besonderen körperlichen Belastungen, kann eine vorübergehende Steigerung der Dosis notwendig werden.
Je nach Dauer der Therapie und angewendeter Dosis ist eine negative Auswirkung auf den Kalziumstoffwechsel zu erwarten. Daher wird eine Osteoporoseprophylaxe empfohlen, insbesondere wenn noch weitere Risikofaktoren vorliegen (einschließlich familiäre Veranlagung, höheres Alter, postmenopausaler Status, unzureichende Protein- und Kalziumzufuhr, übermäßiger Nikotin- und Alkoholkonsum sowie Bewegungsmangel). Die Prophylaxe umfasst eine ausreichende Kalzium- und Vitamin-D-Zufuhr sowie körperliche Betätigung. Bei bereits bestehender Osteoporose sollte eine zusätzliche Therapie in Betracht gezogen werden.
Dieses Arzneimittel enthält Lactose-Monohydrat. Patienten mit der seltenen hereditären Galactoseintoleranz, Lactase-Mangel oder Glucose-Galactose-Malabsorption sollten dieses Arzneimittel nicht einnehmen.
Unter der Behandlung mit Prednisolon in hohen Dosen über einen längeren Zeitraum (30 mg/Tag über mindestens 4 Wochen) sind reversible Störungen der Spermatogenese beobachtet worden, die nach Absetzen des Arzneimittels noch mehrere Monate anhielten.
4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Herzglykoside:
Die Glykosidwirkung kann durch Kaliummangel verstärkt werden.
Saluretika/Laxanzien:
Die Kaliumausscheidung wird verstärkt.
Antidiabetika:
Die blutzuckersenkende Wirkung ist vermindert.
Cumarinderivate:
Die Antikoagulanzienwirkung wird abgeschwächt oder verstärkt.
Nicht-steroidale Entzündungshemmer/Antirheumatika, Salicylate und Indomethacin:
Die Gefahr von Magen-Darm-Blutungen wird erhöht.
Nicht-depolarisierende Muskelrelaxanzien:
Die Muskelrelaxation kann länger anhalten.
Atropin und andere Anticholinergika:
Bei gleichzeitiger Anwendung von Lodotra sind zusätzliche Augeninnendrucksteigerungen möglich. Praziquantel:
Glucocorticoide können die Praziquantel-Konzentration im Blut vermindern.
Chloroquin, Hydroxychloroquin, Mefloquin:
Es besteht ein erhöhtes Risiko des Auftretens von Myopathien, Kardiomyopathien.
Somatropin:
Die Wirksamkeit von Somatropin kann vermindert werden.
Östrogene (z. B. orale Kontrazeptiva):
Die Glucocorticoidwirkung kann verstärkt werden.
Lakritze:
Eine Hemmung der Metabolisierung von Glucocorticoiden ist möglich.
Rifampicin, Phenytoin, Barbiturate, Bupropion, Primidon:
Die Wirksamkeit von Glucocorticoiden ist herabgesetzt.
Ciclosporin:
Die Blutspiegel von Ciclosporin werden erhöht. Es besteht eine erhöhte Gefahr zerebraler Krampfanfälle.
Amphotericin B:
Das Risiko einer Hypokaliämie kann erhöht sein.
Cyclophosphamid:
Die Wirkung von Cyclophosphamid kann verstärkt werden.
ACE-Hemmer:
Erhöhtes Risiko des Auftretens von Blutbildveränderungen.
Aluminium- und magnesiumhaltige Antazida:
Die Resorption von Glucocorticoiden kann herabgesetzt sein. Auf Grund der verzögerten Wirkstofffreisetzung von Lodotra ist jedoch eine Wechselwirkung mit aluminium- und magnesiumhaltigen Antazida unwahrscheinlich.
Beeinflussung von diagnostischen Verfahren:
Hautreaktionen im Rahmen von Allergietests können unterdrückt werden. Der TSH-Anstieg nach Verabreichung von Protirelin kann abgeschwächt werden.
4.6 Fertilität, Schwangerschaft und Stillzeit
Schwangerschaft
Während der Schwangerschaft darf Lodotra nur angewendet werden, wenn die zu erwartenden Vorteile die möglichen Risiken überwiegen. Dabei ist die niedrigste zur Aufrechterhaltung einer adäquaten Krankheitskontrolle erforderliche Lodotra-Dosis anzuwenden.
Tierexperimentelle Studien deuten darauf hin, dass die Verabreichung von Glucocorticoiden während der Schwangerschaft in pharmakologischen Dosen das fetale Risiko für eine intrauterine Wachstumsretardierung, Herz-Kreislauf- oder Stoffwechselerkrankungen im Erwachsenenalter erhöhen kann und möglicherweise eine Auswirkung auf die Glucocorticoid-Rezeptordichte und den Neurotransmitterumsatz oder die Entwicklung des Nervensystems und Verhaltens haben kann.
In tierexperimentellen Studien hat Prednison zur Bildung von Gaumenspalten geführt (siehe Abschnitt 5.3). Die Möglichkeit eines erhöhten Risikos für Gaumenspaltenbildung beim menschlichen Fetus infolge der Verabreichung von Glucocorticoiden während des ersten Trimenons wird derzeit noch diskutiert.
Bei Verabreichung von Glucocorticoiden zum Ende der Schwangerschaft besteht die Gefahr einer Atrophie der Nebennierenrinde beim Fetus, die eine Substitutionsbehandlung des Neugeborenen erforderlich machen kann, die dann langsam reduziert werden muss.
Glucocorticoide gehen in geringen Mengen in die Muttermilch über (bis zu 0,23 % einer Einzeldosis). Bei Dosen bis zu 10 mg täglich liegt die über die Muttermilch aufgenommene Wirkstoffmenge unterhalb der Nachweisgrenze. Eine Schädigung von Säuglingen ist bisher nicht bekannt geworden. Dennoch dürfen Glucocorticoide nur verordnet werden, wenn die Vorteile für Mutter und Kind die Risiken überwiegen.
Da der Milch-/Plasma-Konzentrationsquotient bei Dosen über 10 mg/Tag ansteigt (bei 80 mg Prednison täglich liegt z. B. in der Muttermilch eine Konzentration in Höhe von 25 % des Serumspiegels vor), wird empfohlen, in diesen Fällen mit dem Stillen aufzuhören.
4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Es wurden keine Studien zu den Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen durchgeführt.
4.8 Nebenwirkungen
Die Häufigkeit und Schwere der nachstehend aufgeführten Nebenwirkungen sind von Dosierung und Behandlungsdauer abhängig. In dem für Lodotra empfohlenen Dosisbereich (niedrig dosierte Glucocorticoidtherapie mit Tagesdosen zwischen 1 mg und 10 mg) treten die genannten Nebenwirkungen seltener und mit geringerem Schweregrad auf als bei Dosen über 10 mg.
Folgende Nebenwirkungen können in Abhängigkeit von Behandlungsdauer und Dosis auftreten:
Sehr häufig (> 1/10); häufig (> 1/100 bis < 1/10); gelegentlich (> 1/1.000 bis < 1/100); selten (> 1/10.000 bis < 1/1.000); sehr selten (< 1/10.000); nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
Erkrankungen des Blutes und des Lymphsystems:
Häufig: Mittelgradige Leukozytose, Lymphopenie, Eosinopenie, Polyzythämie
Herzerkrankungen:
Nicht bekannt: Tachykardie
Erkrankungen des Immunsystems
Häufig: Schwächung der Immunabwehr, Maskierung von Infektionen, Exazerbation latenter Infektionen
Selten: Allergische Reaktionen Infektionen und parasitäre Erkrankungen:
Häufig: Erhöhung der Anfälligkeit gegenüber und der Schwere von Infektionen Endokrine Erkrankungen:
Häufig: Adrenale Suppression und Induktion eines Cushing-Syndroms (typische Symptome: Vollmondgesicht, Stammfettsucht und Plethora)
Selten: Störung der Ausschüttung von Sexualhormonen (Amenorrhoe, Impotenz), Störung der Schilddrüsenfunktion
Stoffwechsel- und Ernährungsstörungen:
Häufig: Natriumretention mit Ödembildung, erhöhte Kaliumausscheidung (Achtung: Arrhythmien!), gesteigerter Appetit und Gewichtszunahme, verringerte Glukosetoleranz, Diabetes mellitus, Hypercholesterinämie und Hypertriglyzeridämie
Nicht bekannt: Reversible epidurale, epikardiale oder mediastinale Lipomatose, hypokalämische
Alkalose
Psychiatrische Erkrankungen:
Häufig: Schlaflosigkeit
Selten: Depression, Reizbarkeit, Euphorie, erhöhter Antrieb, Psychose
Erkrankungen des Nervensystems:
Häufig: Kopfschmerzen
Selten: Pseudotumor cerebri, Manifestation einer latenten Epilepsie und erhöhte Anfallsneigung bei manifester Epilepsie
Augenerkrankungen:
Häufig: Katarakt, insbesondere mit subkapsulärer Trübung hinten; Glaukom
Selten: Verschlechterung der Symptomatik verbunden mit Hornhautulzera; Begünstigung von Virus-, Pilz- und bakteriellen Entzündungen des Auges Nicht bekannt: zentrale seröse Chorioretinopathie
Gefäßerkrankungen:
Gelegentlich: Hypertonie, erhöhtes Arteriosklerose- und Thromboserisiko, Vaskulitis (auch als Syndrom nach Beendigung einer Langzeittherapie)
Erkrankungen des Gastrointestinaltrakts:
Gelegentlich (keine gleichzeitige Gabe nichtsteroidaler Entzündungshemmer): Gastrointestinale Ulzera, gastrointestinale Blutungen Selten: Pankreatitis
Nicht bekannt: Übelkeit, Durchfall, Erbrechen Erkrankungen der Haut und des Unterhautzellgewebes:
Häufig: Striae rubrae, Atrophie, Teleangiektasien, erhöhte Kapillarfragilität, Petechien, Ekchymosen
Gelegentlich: Hypertrichose, Steroidakne, verzögerte Wundheilung, Rosacea-ähnliche (periorale)
Dermatitis, Veränderungen der Hautpigmentierung
Selten: Überempfindlichkeitsreaktionen, z. B. Arzneimittelexanthem
Nicht bekannt: Hirsutismus
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen:
Häufig: Muskelatrophie und -schwäche, Osteoporose (dosisabhängig, kann auch bei Kurzzeittherapie auftreten)
Selten: Aseptische Osteonekrose (Humerus- und Femurkopf)
Nicht bekannt: Steroidmyopathie, Sehnenrisse, Frakturen der Wirbel und Röhrenknochen Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung über das Bundesinstitut für Arzneimittel und Medizinprodukte anzuzeigen:
Bundesinstitut für Arzneimittel und Medizinprodukte Abt. Pharmakovigilanz Kurt-Georg-Kiesinger Allee 3 D-53175 Bonn
Website: http://www.bfarm.de/DE/Pharmakovigilanz/form/functions/formpv-node.html
4.9 Überdosierung
Fälle von akuter Intoxikation mit Lodotra sind nicht bekannt. Bei Überdosierung ist mit verstärkten Nebenwirkungen insbesondere auf Endokrinium, Stoffwechsel und Elektrolythaushalt zu rechnen (siehe Abschnitt 4.8).
Ein Antidot für Prednison ist nicht bekannt.
5. PHARMAKOLOGISCHE EIGENSCHAFTEN
5.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Glucocorticoid, ATC-Code: H02AB07 Prednison ist ein nichtfluoriertes Glucocorticoid zur systemischen Therapie.
Prednison beeinflusst dosisabhängig den Stoffwechsel fast aller Gewebe. Im physiologischen Bereich ist diese Wirkung lebensnotwendig zur Aufrechterhaltung der Homöostase des Organismus in Ruhe und unter Belastung sowie zur Regulation der Aktivität des Immunsystems.
Im typischerweise verordneten Dosisbereich von Lodotra wirkt Prednison rasch antiinflammatorisch (antiexsudativ und antiproliferativ) und mit Verzögerung immunsuppressiv. Es hemmt die Chemotaxis und die Aktivität von Zellen des Immunsystems sowie die Freisetzung und Wirkung von Mediatoren der Entzündungs- und Immunreaktionen, z. B. von lysosomalen Enzymen, Prostaglandinen und Leukotrienen.
Die längerfristige Behandlung mit hohen Dosen wirkt dämpfend auf das Immunsystem und die Nebennierenrinde. Der bei Hydrocortison deutlich vorhandene und bei Prednison noch nachweisbare mineralotrope Effekt kann eine Überwachung der Serumelektrolyte erforderlich machen.
Bei Patienten mit rheumatoider Arthritis erreichen die Serumspiegel proinflammatorischer Zytokine wie Interleukin IL-1 und IL-6 und des Tumornekrosefaktors alpha (TNFa) ihre Spitzenwerte in den frühen Morgenstunden (z. B. IL-6 zwischen 7 und 8 Uhr). Nach Einnahme von Lodotra und anschließender Freisetzung von Prednison im Laufe der Nacht (Beginn der Resorption zwischen 2 und 4 Uhr, Cmax zwischen 4 und 6 Uhr) sinken die Zytokinkonzentrationen.
Die Wirksamkeit und Sicherheit von Lodotra wurde in zwei randomisierten, doppelblinden kontrollierten Studien an Patienten mit aktiver rheumatoider Arthritis untersucht.
In der ersten, einer 12-wöchigen multizentrischen, randomisierten, doppelblinden Phase-III-Studie mit insgesamt 288 Patienten, die mit Prednison oder Prednisolon vorbehandelt waren, zeigte die Gruppe, die auf eine vergleichbare Dosis Lodotra umgestellt wurde, eine mittlere Reduktion der Dauer der Morgensteifigkeit um 23 %, während sie in der Vergleichsgruppe unverändert blieb. Einzelheiten sind in der nachstehenden Tabelle dargestellt.
Relative Änderung der Dauer der Morgensteifigkeit nach 12-wöchiger Therapie
|
Relative Änderung [%] |
Lodotra (n = 125) |
Prednison IR (n = 129) |
|
Mittelwert |
-23 |
0 |
|
(SD) |
(89) |
(89) |
|
Median |
-34 |
-13 |
|
(min, max) |
(-100, 500) |
(-100, 610) |
In einer anschließenden, unverblindeten Verlängerungsphase (9 Monate Behandlung) betrug die mittlere relative Veränderung der Dauer der Morgensteifigkeit im Vergleich zu Studienbeginn etwa -
50 %.
Veränderung der Dauer der Morgensteifigkeit nach 12 Monaten Lodotra-Therapie
|
Dauer der Morgensteifigkeit [min] |
Lodotra | |
|
Mittelwert (SD) |
N | |
|
0 Monate Studienbeginn |
156 (97) |
107 |
|
12 Monate Ende der nicht verblindeten Phase |
74 (92) |
96 |
In derselben Studie wurde nach 12 Wochen Therapie in der mit Lodotra behandelten Gruppe eine Abnahme (Median) des proinflammatorischen Zytokins IL-6 um 29 % beobachtet, während in der mit herkömmlichem Prednison behandelten Vergleichsgruppe keine Veränderung zu beobachten war. Auch nach 12 Monaten Therapie mit Lodotra blieb der IL-6-Spiegel unverändert.
Veränderung des IL-6-Spiegels nach 12 Monaten
|
IL-6 [IU/l] |
Loc |
otra |
|
Median (min, max) |
N | |
|
0 Monate |
860 |
142 |
|
Studienbeginn |
(200, 23.000) | |
|
12 Monate Ende der nicht verblindeten Phase |
470 (200, 18.300) |
103 |
Werte <200 IU/l wurden für die statistische Auswertung als 200 IU/l gewertet.
Die Wirksamkeit von Lodotra zusätzlich zu einem DMARD wurde in einer zweiten, randomisierten, Plazebo-kontrollierten Studie an Patienten untersucht, die auf eine alleinige DMARD-Therapie nicht ausreichend ansprachen. Nach 12 Wochen zeigte die Lodotra-Patientengruppe eine signifikant höhere ACR20- und ACR50-Ansprechrate (46,8% bzw. 22,1%) im Vergleich zur Plazebo-Gruppe (29,4% bzw. 10,1%). Die mittlere Abnahme des DAS 28 (disease activity score) vom Ausgangswert (Scores von 5,2 in der Lodotra-Gruppe und 5,1 in der Plazebo-Gruppe) zum Ergebnis nach 12 Wochen war in der Lodotra-Gruppe ebenfalls größer (-1,2 Punkte) als in der Plazebo-Gruppe (-0,7 Punkte).
Außerdem betrug nach 12-wöchiger Behandlung die mittlere Dauer der Morgensteifigkeit in der Lodotra-Gruppe 86,0 Minuten (Abnahme um -66 Minuten) bzw. 114,1 Minuten (Abnahme um -42,6 Minuten) in der Plazebo-Gruppe. Lodotra konnte sicher in Kombination mit DMARDs angewendet werden.
5.2 Pharmakokinetische Eigenschaften
Resorption
Lodotra ist eine Prednison-haltige Tablette mit veränderter Wirkstofffreisetzung. Der Wirkstoff Prednison wird zwischen 4 und 6 Stunden nach Einnahme der Lodotra-Tablette freigesetzt. Danach wird das freigesetzte Prednison rasch und nahezu vollständig resorbiert.
Verteilung
Maximale Plasmaspiegel werden ca. 6 bis 9 Stunden nach der Einnahme erreicht.
Biotransformation
Über 80 % des Prednisons werden bei der ersten Leberpassage in Prednisolon umgewandelt. Das Verhältnis von Prednison zu Prednisolon beträgt etwa 1:6 bis 1:10. Die pharmakologische Wirksamkeit von unverändertem Prednison ist vernachlässigbar. Prednisolon ist der aktive Metabolit. Die Moleküle binden reversibel an Plasmaproteine, mit hoher Affinität für Transcortin (Corticosteroid-bindendes Globulin, CBG) und geringer Affinität zu Plasmaalbumin.
Im Niedrigdosisbereich (bis 5 mg) liegen rund 6 % des Prednisolons ungebunden vor. Die metabolische Elimination verläuft in diesem Bereich dosislinear. Im Dosisbereich oberhalb von 10 mg ist die Bindungskapazität von Transcortin zunehmend ausgeschöpft, und der Anteil von freiem Prednisolon nimmt zu. Dies kann die metabolische Elimination beschleunigen.
Ausscheidung
Die Elimination von Prednisolon erfolgt vornehmlich durch Metabolisierung in der Leber, und zwar zu ca. 70 % durch Glucuronidierung und zu ca. 30 % durch Sulfatierung. Zum Teil erfolgt eine Umwandlung in 11ß,17ß-Dihydroxyandrosta-1,4-dien-3-on und in 1,4-Pregnadien-20-ol. Die Metabolite sind hormonell inaktiv und werden vorwiegend renal eliminiert. Ein vernachlässigbarer Anteil von Prednison und Prednisolon erscheint unverändert im Urin. Die
Plasmaeliminationshalbwertszeit von Prednis(ol)on beträgt ca. 3 Stunden. Bei Patienten mit schweren Leberfunktionsstörungen kann die Halbwertszeit verlängert sein; eine Dosisreduktion sollte in Erwägung gezogen werden. Die biologische Wirkdauer von Prednis(ol)on ist wesentlich länger als die Verweilzeit im Plasma.
Bioverfügbarkeit
In einer Bioverfügbarkeitsstudie mit 27 gesunden Probanden aus dem Jahr 2003 wurden im Vergleich zu Prednison-Tabletten mit sofortiger Wirkstofffreisetzung folgende Ergebnisse ermittelt:
|
Parameter |
Lodotra 5 mg: |
Referenzpräparat 5 mg (Nüchterneinnahme) | |
|
2,5 Stunden nach einer leichten Mahlzeit |
sofort nach einer Mahlzeit | ||
|
Maximale Plasmakonzentration (Cmax): ng/ml |
20,2 (18,5; 21,9) |
21,8 (20,0; 23,7) |
20,7 (19,0; 22,5) |
|
Zeitpunkt der maximalen Plasmakonzentration (Tmax): h |
6,0 (4,5; 10,0) |
6,5 (4,5; 9,0) |
2,0 (1,0; 4,0) |
|
Verzögerung der Wirkstofffreisetzung (tlag): h |
4,0 (3,5; 5,0) |
3,5 (2,0; 5,5) |
0,0 (0,0; 0,5) |
|
Fläche unter der Konzentrations-ZeitKurve (AUC 0-»): ngxh/ml |
110 (101; 119) |
123 (114; 133) |
109 (101; 118) |
Geometrische Kleinste-Quadrate-Mittelwerte und Bereich.
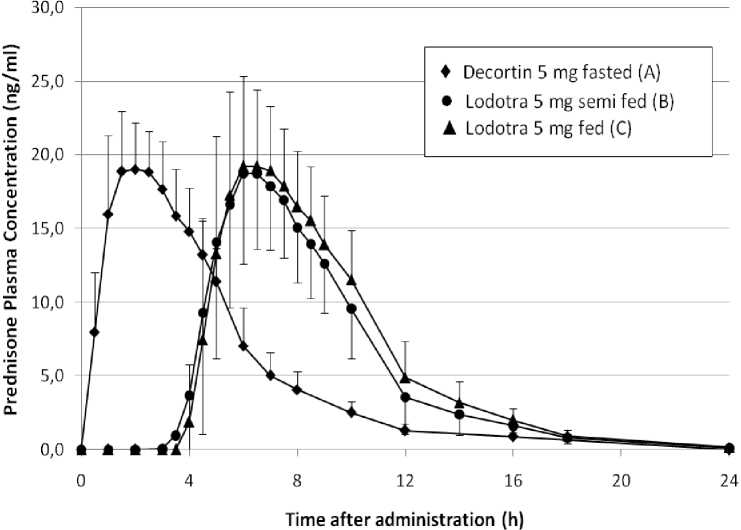
Abbildung: Mittlere Prednison-Plasmaspiegel nach einer Einzeldosis von 5 mg
Prednison, verabreicht als Lodotra 5 mg oder als Tabletten mit sofortiger Wirkstofffreisetzung:
(A) 5-mg-Tabletten mit sofortiger Wirkstofffreisetzung (Einnahme nüchtern um 2 Uhr morgens),
(B) Lodotra 5 mg (2,5 Stunden nach einem leichten Abendessen) und
(C) Lodotra 5 mg (unmittelbar nach einem vollständigen Abendessen).
Die Plasmaspiegelprofile von Lodotra sind vergleichbar mit dem einer Tablette mit sofortiger Wirkstofffreisetzung. Der wesentliche Unterschied besteht darin, dass der Beginn der Wirkstofffreisetzung bei Lodotra nach Einnahme um 4 bis 6 Stunden verzögert ist. Bei 6 % - 7 % der Dosen wurden geringere Plasmakonzentrationen beobachtet.
Dosisproportionalität konnte nach oraler Gabe von Lodotra 1 mg, 2 mg und 5 mg anhand der AUC und Cmax gezeigt werden.
5.3 Präklinische Daten zur Sicherheit
Subchronische/chronische Toxizität
Licht- und elektronenmikroskopische Veränderungen an Langerhans-Inselzellen von Ratten wurde nach täglicher intraperitonealer Gabe von 33 mg/kg KG über 7-14 Tage an Ratten beobachtet. Bei Kaninchen konnten experimentelle Leberschäden durch tägliche Gabe von 2-3 mg/kg KG über 24 Wochen erzeugt werden. Histotoxische Wirkungen (Muskelnekrosen) wurden nach mehrwöchiger Verabreichung von 0,5-5 mg/kg KG bei Meerschweinchen und 4 mg/kg KG bei Hunden beobachtet.
Mutagenes und kanzerogenes Potenzial
Die in tierexperimentellen Studien mit Prednison beobachtete Toxizität stand im Zusammenhang mit überhöhter pharmakologischer Aktivität. In konventionellen Studien zur Genotoxizität waren keine genotoxischen Effekte von Prednison zu erkennen.
Reproduktionstoxizität
In tierexperimentellen Studien zur Reproduktion wurde gezeigt, dass Glucocorticoide wie z. B. Prednison Missbildungen induzieren (Gaumenspalten, Skelettfehlbildungen). Nach parenteraler Gabe wurden geringfügige Anomalien des Schädels, Kiefers und der Zunge bei Ratten festgestellt. Eine intrauterine Retardierung des Wachstums wurde beobachtet (siehe auch Abschnitt 4.6).
Bei Verabreichung therapeutischer Dosen werden vergleichbare Effekte bei Patienten als unwahrscheinlich erachtet.
6. PHARMAZEUTISCHE ANGABEN
6.1 Liste der sonstigen Bestandteile
Tablettenkern:
Hochdisperses Siliciumdioxid Croscarmellose-Natrium Lactose-Monohydrat Magnesiumstearat (Ph. Eur.) [pflanzlich]
Povidon K 29/32 Eisen(III)-oxid (E 172)
Tablettenmantel:
Hochdisperses Siliciumdioxid Calciumhydrogenphosphat-Dihydrat Glyceroldibehenat (Ph. Eur.)
Magnesiumstearat (Ph. Eur.) [pflanzlich]
Povidon K 29/32
Eisen(III)-hydroxid-oxid x H2O (E 172)
6.2 Inkompatibilitäten Nicht zutreffend
6.3 Dauer der Haltbarkeit
2 Jahre
Haltbarkeit nach dem ersten Öffnen der Flaschen: 14 Wochen.
6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung Nicht über 25 °C lagern.
6.5 Art und Inhalt des Behältnisses
Behältnis mit 30 und 100 Tabletten mit veränderter Wirkstofffreisetzung Weiße Flasche aus Polyethylen hoher Dichte (HDPE).
HDPE-Schraubverschluss (mit einer Trockenmittelkapsel) mit drei erhöhten Punkten am Flaschenrand, um das Öffnen des Behälters zu erleichtern.
Behältnis mit 500 Tabletten mit veränderter Wirkstofffreisetzung
Weiße Flasche aus Polyethylen hoher Dichte (mit einem geringen LDPE-Anteil). Polypropylen-Schraubverschluss (ohne drei erhöhte Punkte).
Packungsgrößen: Flaschen mit 30 und 100 Tabletten mit veränderter
Wirkstofffreisetzung
Klinikpackungen: Flaschen mit 500 Tabletten mit veränderter
Wirkstofffreisetzung
6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung
Keine besonderen Anforderungen.
7. INHABER DER ZULASSUNG
Horizon Pharma GmbH Joseph-Meyer-Str. 13-15 68167 Mannheim
Mitvertrieb Mundipharma GmbH Mundipharma Straße 2 65549 Limburg Telefon: (0 64 31) 701-0 Telefax: (0 64 31) 7 42 72
8. ZULASSUNGSNUMMER(N)
Lodotra 1 mg Tabletten mit veränderter Wirkstofffreisetzung
67040.00. 00
Lodotra 2 mg Tabletten mit veränderter Wirkstofffreisetzung
67041.00. 00
Lodotra 5 mg Tabletten mit veränderter Wirkstofffreisetzung
67042.00. 00
9. DATUM DER ERTEILUNG DER ZULASSUNG/VERLÄNGERUNG DER ZULASSUNG
Datum der Erteilung der Zulassung: 16.03.2009/04.03.2014
10. STAND DER INFORMATION
August 2014
11. VERKAUFSABGRENZUNG V erschreibungspflichtig
Weitere Angaben:
Mundipharma Service für Fragen zum Präparat und zur Therapie:
- Gebührenfreie Info-Line: (0800) 8 55 11 11
- E-Mail: medinfo@mundipharma.de
- Internet: http://www.mundipharma.de