Novalgin 1 G-Injektionslösung
FACHINFORMATION
1. BEZEICHNUNG DER ARZNEIMITTEL
Novalgin® 1 g-Injektionslösung Novalgin® 2,5 g-Injektionslösung
Wirkstoff: Metamizol-Natrium 1 H2O
2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
Novalgin 1 g-Injektionslösung
1 ml Injektionslösung enthält 500 mg Metamizol-Natrium 1 H2O.
Jede Ampulle mit 2 ml Injektionslösung enthält 1 g Metamizol-Natrium 1 H2O.
Novalgin 2,5 g-Injektionslösung
1 ml Injektionslösung enthält 500 mg Metamizol-Natrium 1 H2O.
Jede Ampulle mit 5 ml Injektionslösung enthält 2,5 g Metamizol-Natrium 1 H2O.
Vollständige Auflistung der sonstigen Bestandteile siehe Abschnitt 6.1.
3. DARREICHUNGSFORM
Injektionslösung, klar, nahezu farblos bis gelb.
4. KLINISCHE ANGABEN
4.1 Anwendungsgebiete
- Akute starke Schmerzen nach Verletzungen oder Operationen,
- Koliken,
- Tumorschmerzen,
- sonstige akute oder chronische starke Schmerzen, soweit andere therapeutische Maßnahmen nicht indiziert sind,
- hohes Fieber, das auf andere Maßnahmen nicht anspricht.
Die parenterale Anwendung ist nur indiziert, sofern eine enterale Applikation nicht infrage kommt.
4.2 Dosierung und Art der Anwendung Dosierung
Die Dosierung richtet sich nach der Intensität der Schmerzen oder des Fiebers und der individuellen Empfindlichkeit, auf Novalgin zu reagieren.
Grundsätzlich sollte die niedrigste schmerz- und fieberkontrollierende Dosis gewählt werden.
Bei Fieber ist für Kinder eine Dosis von 10 mg Metamizol pro Kilogramm Körpergewicht im Allgemeinen ausreichend.
30 bis 60 Minuten nach oraler und etwa 30 Minuten nach parenteraler Gabe kann eine deutliche Wirkung erwartet werden.
Für Kinder und Jugendliche bis 14 Jahre gilt, dass als Einzeldosis 8 bis 16 mg Metamizol-Natrium 1 H2O pro Kilogramm Körpergewicht gegeben werden. Erwachsene und Jugendliche ab 15 Jahren (> 53 kg) können bis zu 1000 mg pro Einzeldosis einnehmen. Bei unzureichender Wirkung kann die jeweilige Einzeldosis, in Abhängigkeit von der Tagesmaximaldosis, bis zu 4-mal am Tag gegeben werden.
Die folgende Dosierungstabelle enthält die empfohlenen Einzeldosen und maximalen Tagesdosen.
Bei parenteraler Applikation werden üblicherweise als Einzeldosis 6 mg bis 16 mg Metamizol pro Kilogramm Körpergewicht gegeben. Säuglinge ab dem 3. Lebensmonat bis zu 1 Jahr erhalten Novalgin Injektionslösung ausschließlich intramuskulär.
Da hypotensive Reaktionen auf die Injektion möglicherweise dosisabhängig sind, muss die Indikation für parenterale Einzeldosen von mehr als 1 g Novalgin streng gestellt werden.
|
Alter (Körpergewicht) |
Einzeldosis |
|
3-11 Monate (5-8 kg) |
0,1-0,2 ml Novalgin (entsprechend 50-100 mg Metamizol-Natrium 1 H2O) nur i. m. |
|
1-3 Jahre (9-15 kg) |
0,2-0,5 ml Novalgin (entsprechend 100-250 mg Metamizol-Natrium 1 H2O) |
|
4-6 Jahre (16-23 kg) |
0,3-0,8 ml Novalgin (entsprechend 150-400 mg Metamizol-Natrium 1 H2O) |
|
7-9 Jahre (24-30 kg) |
0,4-1 ml Novalgin (entsprechend 200-500 mg Metamizol-Natrium 1 H2O) |
|
10-12 Jahre (31-45 kg) |
0,5-1 ml Novalgin (entsprechend 250-500 mg Metamizol-Natrium 1 H2O) |
|
13-14 Jahre (46-53 kg) |
0,8-1,8 ml Novalgin (entsprechend 400-900 mg Metamizol-Natrium 1 H2O) |
|
Erwachsene und Jugendliche ab 15 Jahren (> 53 kg) |
1-2 ml*) Novalgin (entsprechend 500-1000 mg Metamizol-Natrium 1 H2O) |
*) Bei Bedarf kann die Einzeldosis auf 5 ml (entsprechend 2500 mg Metamizol-Natrium 1 H2O) und die Tagesdosis auf 10 ml (entsprechend 5000 mg Metamizol-Natrium 1 H2O) erhöht werden.
Ältere Patienten
Bei älteren Patienten sollte die Dosis vermindert werden, da die Ausscheidung der Stoffwechselprodukte von Novalgin verzögert sein kann.
Bei reduziertem Allgemeinzustand und eingeschränkter Kreatininclearance
Bei Patienten mit reduziertem Allgemeinzustand und eingeschränkter Kreatininclearance sollte die Dosis vermindert werden, da die Ausscheidung der Stoffwechselprodukte von Novalgin verzögert sein kann.
Eingeschränkte Nieren- oder Leberfunktion
Da bei eingeschränkter Nieren- oder Leberfunktion die Eliminationsgeschwindigkeit vermindert ist, sollten mehrfache hohe Dosen vermieden werden. Bei nur kurzzeitiger Anwendung ist keine Dosisreduktion notwendig. Zur Langzeitanwendung liegen keine Erfahrungen vor.
Art der Anwendung
Die Applikationsart richtet sich nach dem gewünschten therapeutischen Effekt und dem Zustand des Patienten. In vielen Fällen ist die orale Gabe ausreichend, um eine zufriedenstellende Wirkung zu erzielen. Ist ein schnell einsetzender Effekt erforderlich oder ist die orale bzw. rektale Gabe nicht indiziert, wird die intravenöse oder intramuskuläre Injektion von Novalgin empfohlen. Bei der Wahl der Applikationsweise ist zu bedenken, dass die parenterale Medikamentengabe mit einem höheren Risiko anaphylaktischer bzw. anaphylaktoider Reaktionen verbunden ist.
Novalgin Injektionslösung wird intravenös oder intramuskulär - bei Säuglingen (3-11 Monate) ausschließlich intramuskulär - injiziert. Die intramuskuläre Injektion sollte stets mit körperwarmer Lösung vorgenommen werden.
Novalgin Injektionslösung kann mit 5%iger Glukose-, 0,9%iger Kochsalz- oder Ringer-Laktat-Lösung gemischt bzw. verdünnt werden. Da solche Mischungen allerdings nur begrenzt stabil sind, müssen sie sofort infundiert werden.
Wegen der Möglichkeit von Inkompatibilitäten wird empfohlen, Novalgin Injektionslösung nicht zusammen mit anderen Medikamenten zu injizieren oder zu infundieren.
Dauer der Anwendung
Die Dauer der Anwendung richtet sich nach Art und Schwere der Erkrankung. Bei längerfristiger Therapie mit Novalgin sind regelmäßige Blutbildkontrollen einschließlich Differenzialblutbild erforderlich.
Sicherheitsvorkehrungen bei der Injektion
Eine Einzeldosis von mehr als 2 ml Novalgin (entsprechend 1000 mg Metamizol-Natrium 1 H2O) bedarf einer besonders sorgfältigen Indikationsstellung, da der Verdacht besteht, dass der nicht allergisch bedingte kritische Blutdruckabfall von der Dosis abhängt.
Die parenterale Gabe von Novalgin muss beim liegenden Patienten und unter sorgfältiger ärztlicher Überwachung erfolgen.
Um die Gefahr einer hypotensiven Reaktion zu minimieren und um sicherzustellen, dass die Injektion bei den ersten Zeichen einer anaphylaktischen bzw. anaphylaktoiden Reaktion abgebrochen werden kann, darf die intravenöse Injektion nur sehr langsam erfolgen, d. h. nicht schneller als 1 ml (entsprechend 500 mg Metamizol-Natrium 1 H2O) pro Minute.
4.3 Gegenanzeigen
- Überempfindlichkeit gegen den Wirkstoff oder andere Pyrazolone bzw. Pyrazolidine (dies schließt auch Patienten ein, die z. B. mit einer Agranulozytose nach Anwendung dieser Substanzen reagiert haben) oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile von Novalgin,
- Patienten mit bekanntem Analgetika-Asthma-Syndrom oder bekannter Analgetika-Intoleranz vom Urtikaria-Angioödemtyp, d. h. Patienten, die mit Bronchospasmus oder anderen anaphylaktoiden Reaktionsformen (z. B. Urtikaria, Rhinitis, Angioödem) auf Salicylate, Paracetamol oder andere nicht narkotische Analgetika wie z. B. Diclofenac, Ibuprofen, Indometacin oder Naproxen reagieren,
- Störungen der Knochenmarkfunktion (z. B. nach Zytostatikabehandlung) oder Erkrankungen des hämatopoetischen Systems,
- genetisch bedingter Glukose-6-Phosphat-Dehydrogenasemangel (Hämolysegefahr),
- akute intermittierende hepatische Porphyrie (Gefahr der Auslösung einer Porphyrie-Attacke),
- letztes Drittel der Schwangerschaft (siehe Abschnitt 4.6),
- Stillzeit (siehe Abschnitt 4.6),
- Neugeborene und Säuglinge unter 3 Monaten oder unter 5 kg Körpergewicht, da kein wissenschaftliches Erkenntnismaterial über die Anwendung vorliegt,
- Säuglinge (3-11 Monate) als intravenöse Injektion,
- bestehende Hypotonie und instabile Kreislaufsituation.
4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Novalgin enthält das Pyrazolonderivat Metamizol und besitzt die seltenen, aber lebensbedrohlichen Risiken des Schocks und der Agranulozytose (siehe Abschnitt 4.8).
Patienten, die auf Novalgin anaphylaktoide Reaktionen zeigen, sind auch besonders gefährdet, in gleicher Weise auf andere nicht narkotische Analgetika zu reagieren.
Patienten, die auf Novalgin eine anaphylaktische oder eine andere immunologisch vermittelte Reaktion (z. B. Agranulozytose) zeigen, sind auch besonders gefährdet, in gleicher Weise auf andere Pyrazolone und Pyrazolidine zu reagieren.
Agranulozytose
Wenn Zeichen einer Agranulozytose oder Thrombozytopenie (siehe Abschnitt 4.8) auftreten, muss sofort die Anwendung von Novalgin abgebrochen und das Blutbild (einschließlich Differenzialblutbild) kontrolliert werden. Mit dem Abbruch der Behandlung darf nicht gewartet werden, bis die Ergebnisse der Laboruntersuchungen vorliegen.
Panzytopenie
Bei Auftreten einer Panzytopenie muss die Behandlung sofort abgebrochen werden und das komplette Blutbild überwacht werden, bis es sich normalisiert (siehe Abschnitt 4.8). Alle Patienten sollten darauf hingewiesen werden, dass sie sofort den Arzt aufsuchen sollten, wenn während der Behandlung Krankheitszeichen und Symptome auftreten, die auf eine Blutdyskrasie hindeuten (z. B. allgemeines Unwohlsein, Infektion, anhaltendes Fieber, Blutergüsse, Blutungen, Blässe).
Anaphylaktische/anaphylaktoide Reaktionen
Bei der Wahl der Applikationsweise ist zu bedenken, dass die parenterale Gabe von Novalgin mit einem höheren Risiko anaphylaktischer bzw. anaphylaktoider Reaktionen verbunden ist (siehe Abschnitt 4.2, „Sicherheitsvorkehrungen bei der Injektion“).
Die Gefahr möglicherweise schwerer anaphylaktoider Reaktionen auf Novalgin ist deutlich erhöht für Patienten mit:
- Analgetika-Asthma-Syndrom oder Analgetika-Intoleranz vom Urtikaria-Angioödemtyp (siehe Abschnitt 4.3),
- Asthma bronchiale, insbesondere mit gleichzeitig bestehender Rhinosinusitis und Nasenpolypen,
- chronischer Urtikaria,
- Intoleranz gegenüber Farbstoffen (z. B. Tartrazin) bzw. Konservierungsmitteln (z. B. Benzoate),
- Alkoholintoleranz. Solche Patienten reagieren schon auf geringe Mengen an alkoholischen Getränken mit Symptomen wie Niesen, Augentränen und starker Gesichtsrötung. Eine solche Alkoholintoleranz kann ein Hinweis auf ein bisher nicht diagnostiziertes Analgetika-Asthma-Syndrom sein (siehe Abschnitt 4.3).
Zu einem anaphylaktischen Schock kann es vorwiegend bei empfindlichen Patienten kommen. Daher ist besondere Vorsicht bei der Anwendung bei Patienten mit Asthma oder Atopie geboten.
Schwere Hautreaktionen
Die lebensbedrohlichen Hautreaktionen Stevens-Johnson-Syndrom (SJS) und toxische epidermale Nekrolyse (TEN) wurden bei Verwendung von Metamizol berichtet. Falls sich Symptome oder Zeichen eines SJS oder einer TEN entwickeln (wie progressiver Hautausschlag, oft mit Blasen oder Läsionen der Schleimhaut), muss die Behandlung mit Novalgin sofort abgebrochen werden und darf zu keiner Zeit neu eingeführt werden.
Patienten sollten auf die Zeichen und Symptome aufmerksam gemacht werden und engmaschig auf Hautreaktionen überwacht werden, insbesondere in den ersten Wochen der Behandlung.
Isolierte hypotensive Reaktionen
Novalgin kann hypotensive Reaktionen auslösen (siehe auch Abschnitt 4.8). Diese Reaktionen sind möglicherweise dosisabhängig. Hiermit ist bei parenteraler Gabe eher zu rechnen als bei enteraler.
Die Gefahr solcher Reaktionen ist ebenfalls erhöht bei:
- zu schneller intravenöser Injektion (siehe Abschnitt 4.2),
- Patienten mit z. B. vorbestehender Hypotonie, Volumenmangel oder Dehydratation, instabilem Kreislauf oder beginnendem Kreislaufversagen (wie z. B. bei Patienten mit Herzinfarkt oder Polytrauma),
- Patienten mit hohem Fieber.
Deshalb sind sorgfältige Indikationsprüfung und engmaschige Überwachung bei diesen Patienten erforderlich. Vorbeugende Maßnahmen (z. B. Kreislaufstabilisierung) können nötig sein, um das Risiko von hypotensiven Reaktionen zu reduzieren.
Novalgin darf nur unter sorgfältiger Überwachung der hämodynamischen Parameter eingesetzt werden bei Patienten, bei denen eine Senkung des Blutdrucks auf jeden Fall vermieden werden muss, wie z. B. bei schwerer koronarer Herzkrankheit oder relevanten Stenosen der hirnversorgenden Gefäße.
Novalgin sollte nur nach strenger Nutzen-Risiko-Abwägung und entsprechenden Vorsichtsmaßnahmen angewendet werden bei Patienten mit Nieren- oder Leberfunktionsstörungen (siehe Abschnitt 4.2).
Vor der Gabe von Novalgin muss der Patient entsprechend befragt werden. Bei Patienten mit erhöhtem Risiko für anaphylaktoide Reaktionen darf Novalgin nur nach sorgfältiger Abwägung möglicher Risiken gegen den erwarteten Nutzen eingesetzt werden. Wird Novalgin in solchen Fällen gegeben, ist der Patient engmaschig ärztlich zu überwachen und Notfallbereitschaft sicherzustellen.
Auf äußerer Umhüllung:
Warnhinweis: Enthält Metamizol.
1 ml enthält 1,42 mmol (32,7 mg) Natrium. Dies ist zu berücksichtigen bei Personen unter natriumkontrollierter (natriumarmer/kochsalzarmer) Diät.
4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Metamizol kann eine Abnahme der Ciclosporin-Serumspiegel bewirken. Diese müssen daher überwacht werden, wenn gleichzeitig Novalgin angewendet wird.
Bei gleichzeitiger Anwendung von Novalgin und Chlorpromazin kann eine schwere Hypothermie auftreten.
Die zusätzliche Gabe von Metamizol zu Methotrexat kann die Hämatotoxizität von Methotrexat verstärken, insbesondere bei älteren Patienten. Diese Kombination sollte deshalb vermieden werden.
Metamizol kann bei gleichzeitiger Anwendung die Wirkung von Acetylsalicylsäure auf die Thrombozytenaggregation vermindern. Daher sollte Metamizol bei Patienten, die Acetylsalicylsäure in niedriger Dosierung zur Kardioprotektion einnehmen, mit Vorsicht angewendet werden.
Bupropion-Blutspiegel können durch Metamizol erniedrigt werden. Daher ist bei gleichzeitiger Anwendung von Metamizol und Bupropion Vorsicht geboten.
Für die Substanzklasse der Pyrazolone ist bekannt, dass es zu Wechselwirkungen mit oralen Antikoagulanzien, Captopril, Lithium und Triamteren sowie Änderungen der Wirksamkeit von Antihypertensiva und Diuretika kommen kann. Inwieweit auch Metamizol zu diesen Wechselwirkungen führt, ist nicht bekannt.
4.6 Schwangerschaft und Stillzeit
Schwangerschaft
Es liegen keine hinreichenden Daten für die Verwendung von Novalgin bei Schwangeren vor. Metamizol ist plazentagängig. In tierexperimentellen Studien zeigte Metamizol keine teratogenen Effekte (siehe Abschnitt 5.3). Da keine hinreichenden Erfahrungen für den Menschen vorliegen, sollte Novalgin im ersten Trimenon nicht und im zweiten Trimenon nur nach strenger ärztlicher NutzenRisiko-Abwägung eingenommen/angewendet werden.
Obwohl Metamizol ein nur schwacher Prostaglandinsynthese-Hemmer ist, können die Möglichkeit eines vorzeitigen Verschlusses des Ductus arteriosus (Botalli) sowie perinatale Komplikationen infolge einer Reduktion der kindlichen und mütterlichen Thrombozytenaggregabilität nicht ausgeschlossen werden. Novalgin ist daher während des letzten Trimenons der Schwangerschaft kontraindiziert (siehe Abschnitt 4.3).
Stillzeit
Die Metaboliten von Metamizol werden in die Muttermilch ausgeschieden, daher darf während der Einnahme/Anwendung und bis zu mindestens 48 Stunden nach der letzten Einnahme/Anwendung von Novalgin nicht gestillt werden (siehe Abschnitt 4.3).
4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Im empfohlenen Dosisbereich ist keine Beeinträchtigung des Konzentrations- und Reaktionsvermögens bekannt. Vorsichtshalber sollte aber, zumindest bei höheren Dosierungen, die Möglichkeit einer Beeinträchtigung in Betracht gezogen werden und auf das Bedienen von
Maschinen, das Führen von Fahrzeugen oder sonstige gefahrvolle Tätigkeiten verzichtet werden. Dies gilt besonders im Zusammenwirken mit Alkohol.
4.8 Nebenwirkungen
Bei den Häufigkeitsangaben zu Nebenwirkungen werden folgende Kategorien zugrunde gelegt:
Sehr häufig (> 1/10)
Häufig (> 1/100. < 1/10)
Gelegentlich (> 1/1.000, < 1/100)
Selten (> 1/10.000, < 1/1.000)
Sehr selten (< 1/10.000)
Nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar)
Erkrankungen des Blutes und des Lymphsystems Selten: Leukopenie.
Sehr selten: Agranulozytose, einschließlich Fälle mit tödlichem Ausgang, Thrombozytopenie.
Häufigkeit
nicht bekannt: Aplastische Anämie, Panzytopenie, einschließlich Fälle mit tödlichem Ausgang.
Diese Reaktionen können auch auftreten, wenn Metamizol bei früheren Gelegenheiten ohne Komplikationen gegeben wurde.
Es gibt vereinzelt Hinweise, wonach das Risiko einer Agranulozytose möglicherweise erhöht sein kann, wenn Novalgin länger als eine Woche angewendet wird.
Diese Reaktion ist nicht dosisabhängig und kann zu jedem Zeitpunkt der Behandlung auftreten. Sie äußert sich in hohem Fieber, Schüttelfrost, Halsschmerzen, Schluckbeschwerden sowie Entzündung im Mund-, Nasen-, Rachen- und Genital- oder Analbereich. Bei Patienten, die Antibiotika erhalten, können diese Zeichen allerdings minimal sein. Lymphknoten- oder Milzschwellung ist gering oder fehlt ganz. Die Blutsenkung ist stark beschleunigt, die Granulozyten sind erheblich vermindert oder fehlen vollständig. Im Allgemeinen, aber nicht immer, finden sich normale Werte für Hämoglobin, Erythrozyten und Thrombozyten (siehe Abschnitt 4.4).
Für die Heilung ist das sofortige Absetzen entscheidend. Daher wird dringend empfohlen, Novalgin sofort abzusetzen und nicht erst die Ergebnisse der labordiagnostischen Untersuchungen abzuwarten, wenn es zu einer unerwarteten Verschlechterung des Allgemeinbefindens kommt, das Fieber nicht abklingt oder neu auftritt oder schmerzhafte Schleimhautveränderungen besonders im Mund-, Nasen- und Rachenraum auftreten.
Bei Auftreten einer Panzytopenie muss die Behandlung sofort abgebrochen werden und das komplette Blutbild überwacht werden, bis es sich normalisiert (siehe Abschnitt 4.4).
Erkrankungen des Immunsystems
Selten: Anaphylaktoide oder anaphylaktische Reaktionen*.
Sehr selten: Analgetika-induziertes Asthma-Syndrom.
Bei Patienten mit Analgetika-Asthma-Syndrom manifestieren sich Unverträglichkeitsreaktionen typischerweise in Form von Asthmaanfällen.
Häufigkeit
nicht bekannt: Anaphylaktischer Schock*.
*Diese Reaktionen können insbesondere nach parenteraler Applikation auftreten, schwerwiegend und lebensbedrohlich sein, in manchen Fällen sogar mit tödlichem Ausgang. Sie können auch auftreten, wenn Metamizol bei früheren Gelegenheiten ohne Komplikationen gegeben wurde.
Solche Reaktionen können sich während der Injektion bzw. unmittelbar nach der Einnahme, aber auch Stunden später entwickeln. Sie treten allerdings überwiegend während der ersten Stunde nach Gabe auf. Leichtere Reaktionen manifestieren sich typischerweise in Haut- und Schleimhautreaktionen (wie z. B. Juckreiz, Brennen, Rötung, Urtikaria, Schwellungen), Dyspnoe und - seltener -gastrointestinalen Beschwerden. Solche leichteren Reaktionen können in schwerere Formen übergehen mit generalisierter Urtikaria, schweren Angioödemen (auch im Larynxbereich), schwerem Bronchospasmus, Herzrhythmusstörungen, Blutdruckabfall (manchmal auch mit vorausgehendem Blutdruckanstieg), Kreislaufschock.
Daher ist Novalgin bei Auftreten von Hautreaktionen sofort abzusetzen.
Herzerkrankungen
Häufigkeit
nicht bekannt: Kounis-Syndrom.
Gefäßerkrankungen
Gelegentlich: Hypotensive Reaktionen während oder nach der Anwendung, die möglicherweise
pharmakologisch bedingt und nicht von anderen Zeichen einer anaphylaktoiden bzw. anaphylaktischen Reaktion begleitet sind. Eine solche Reaktion kann bis zu einem schweren Blutdruckabfall führen. Schnelle intravenöse Injektion erhöht das Risiko einer hypotensiven Reaktion.
Auch bei Hyperpyrexie kann es dosisabhängig zu einem kritischen Blutdruckabfall ohne weitere Anzeichen einer Überempfindlichkeitsreaktion kommen.
Erkankungen der Haut und des Unterhautzellgewebes Gelegentlich: Fixes Arzneimittelexanthem.
Selten: Ausschlag (z. B. makulopapulöses Exanthem).
Sehr selten: Stevens-Johnson-Syndrom oder toxische epidermale Nekrolyse (Behandlung abbrechen, siehe Abschnitt 4.4).
Erkrankungen der Nieren und Harnwege
Sehr selten: Akute Verschlechterung der Nierenfunktion, wobei sich sehr selten eine Proteinurie,
Oligo- oder Anurie bzw. ein akutes Nierenversagen entwickeln kann, akute interstitielle Nephritis.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Bei Injektionen können Schmerzen an der Einstichstelle und lokale Reaktionen, sehr selten bis hin zu Phlebitiden, auftreten.
Über eine Rotfärbung des Urins ist berichtet worden, die auf dem harmlosen, in geringer Konzentration vorliegenden Metamizol-Metaboliten Rubazonsäure beruhen kann.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem
Bundesinstitut für Arzneimittel und Medizinprodukte Abt. Pharmakovigilanz Kurt-Georg-Kiesinger-Allee 3 D-53175 Bonn Website: www.bfarm.de
anzuzeigen.
4.9 Überdosierung
Symptome einer Überdosierung:
Im Rahmen akuter Überdosierungen wurden Übelkeit, Erbrechen, Schmerzen im Abdominalbereich, eine Einschränkung der Nierenfunktion/akutes Nierenversagen (z. B. unter dem Bild einer interstitiellen Nephritis) und - seltener - zentralnervöse Symptome (Schwindel, Somnolenz, Koma, Krämpfe) und Blutdruckabfall bis hin zum Schock und Tachykardie beobachtet.
Nach sehr hohen Dosen kann die Ausscheidung von Rubazonsäure eine Rotverfärbung des Urins verursachen.
Therapiemaßnahmen bei Überdosierung:
Für Metamizol ist kein spezifisches Antidot bekannt. Liegt die Einnahme von Metamizol nur kurz zurück, kann versucht werden, die Aufnahme in den Körper durch Maßnahmen der primären Detoxifikation (z. B. Magenspülung) oder resorptionsmindernde Maßnahmen (z. B. Aktivkohle) zu begrenzen. Der Hauptmetabolit (4-N-Methylaminoantipyrin) kann durch Hämodialyse, Hämofiltration, Hämoperfusion oder Plasmafiltration eliminiert werden.
Die Behandlung der Intoxikation kann, ebenso wie die Prävention von schweren Komplikationen, allgemeine und spezielle intensivmedizinische Überwachung und Behandlung erforderlich machen.
Sofortmaßnahmen bei schweren Überempfindlichkeitsreaktionen (Schock):
Bei den ersten Anzeichen (z. B. kutane Reaktionen wie Urtikaria und Flush, Unruhe, Kopfschmerz, Schweißausbruch, Übelkeit) Injektion abbrechen. Kanüle in der Vene belassen oder einen venösen Zugang schaffen. Neben gebräuchlichen Notfallmaßnahmen wie Kopf-Oberkörper-Tieflage, Atemwege freihalten, Applikation von Sauerstoff kann die Gabe von Sympathomimetika, Volumen oder Glukokortikoiden notwendig werden.
5. PHARMAKOLOGISCHE EIGENSCHAFTEN
5.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Analgetikum, Antipyretikum,
ATC-Code: N02B B02.
Metamizol ist ein Pyrazolonderivat und hat analgetische, antipyretische und spasmolytische Eigenschaften. Der Wirkungsmechanismus ist nicht vollständig aufgeklärt. Einige Untersuchungsergebnisse zeigen, dass Metamizol und der Hauptmetabolit (4-N-Methylaminoantipyrin) vermutlich sowohl einen zentralen als auch einen peripheren Wirkungsmechanismus haben.
5.2 Pharmakokinetische Eigenschaften
Metamizol wird nach oraler Applikation vollständig zum pharmakologisch wirksamen 4-N-Methyl-aminoantipyrin (MAA) hydrolysiert. Die Bioverfügbarkeit von MAA liegt bei ca. 90 % und ist nach oraler Gabe etwas höher als nach parenteraler Gabe. Die gleichzeitige Einnahme von Mahlzeiten hat keinen relevanten Einfluss auf die Kinetik von Metamizol.
Die klinische Wirksamkeit beruht hauptsächlich auf MAA, zu einem gewissen Ausmaß auch auf dem Metaboliten 4-Aminoantipyrin (AA). Die AUC-Werte für AA bilden ca. 25 % der AUC-Werte für MAA. Die Metaboliten 4-N-Acetylaminoantipyrin (AAA) und 4-N-Formylaminoantipyrin (FAA) sind anscheinend pharmakologisch inaktiv.
Zu beachten ist, dass alle Metaboliten eine nicht lineare Pharmakokinetik besitzen. Eine klinische Bedeutung dieses Phänomens ist nicht bekannt. Bei einer Kurzzeitbehandlung ist die Akkumulation der Metaboliten von geringer Bedeutung.
Metamizol ist plazentagängig. Die Metaboliten von Metamizol werden in die Muttermilch ausgeschieden.
Die Plasmaproteinbindung beträgt für MAA 58 %, für AA 48 %, für FAA 18 % und für AAA 14 %.
Nach intravenöser Applikation beträgt die Plasmahalbwertszeit für Metamizol ca. 14 Minuten. Etwa 96 % einer radioaktiv markierten Dosis werden nach intravenöser Gabe im Urin und etwa 6 % in den Faeces wiedergefunden. Nach einer oralen Einzeldosis konnten 85 % der im Urin ausgeschiedenen Metaboliten identifiziert werden. Davon waren 3±1 % MAA, 6±3 % AA, 26±8 % AAA und 23±4 % FAA. Die renale Clearance nach einer oralen Einzeldosis von 1 g Metamizol betrug für MAA 5±2, für AA 38±13, für AAA 61±8 und für FAA 49±5 ml/min. Die zugehörigen Plasmahalbwertszeiten waren 2,7±0,5 Stunden für MAA, 3,7±1,3 Stunden für AA, 9,5±1,5 Stunden für AAA und 11,2± 1,5 Stunden für FAA.
Ältere Menschen
Bei der Behandlung älterer Patienten erhöht sich die AUC auf das 2- bis 3-Fache. Nach oraler Einzelgabe stieg bei Patienten mit Leberzirrhose die Halbwertszeit von MAA und FAA etwa auf das 3-Fache, während die Halbwertszeit von AA und AAA nicht in demselben Maß anstieg. Bei diesen Patienten sollten hohe Dosen vermieden werden.
Nierenfunktionsstörungen
Die verfügbaren Daten von Patienten mit eingeschränkter Nierenfunktion zeigen eine verminderte Eliminationsgeschwindigkeit für einige Metaboliten (AAA und FAA). Deshalb sollten bei diesen Patienten hohe Dosen vermieden werden.
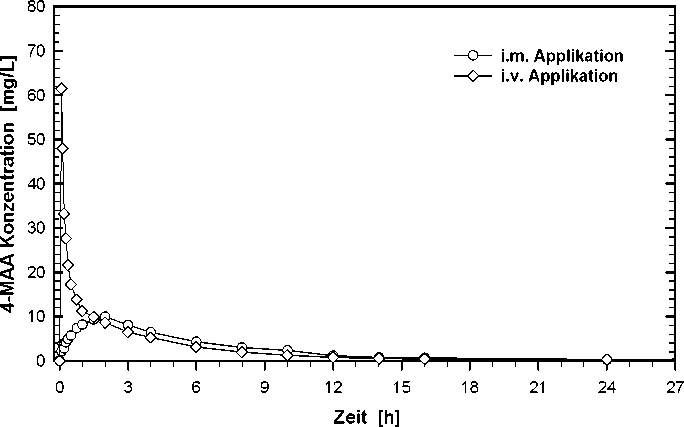
Bioverfügbarkeit
Eine im Jahre 1989 durchgeführte Bioverfügbarkeitsuntersuchung mit der i.m. Lösung an 12 Probanden ergab im Vergleich zum Referenzpräparat (i. v. Applikation in 2 Minuten) für 4-MAA:
i. m. i. v.
Applikation Applikation
(1 g) (1 g)
maximale Plasmakonzentration (Cmax) [mg/L] 11,4 ± 3,12 62,1 ± 15,9
Zeitpunkt der maximalen Plasmakonzentration (tmax) [h] 1,67 ± 0,69 0,09 ± 0,02
Fläche unter der Konzentrations-Zeit-Kurve (AUC) [mg x h/L] 64,1 ±14,8 67,8 ± 16,1
(Angabe der Werte als Mittelwert und Standardabweichung)
Die absolute Bioverfügbarkeit der i. m. Lösung, gemessen an der AUC für die 4-MAA-Plasmakonzentrationen, beträgt 87 %.
Mittlere Plasmaspiegelverläufe im Vergleich zu einem Referenzpräparat in einem KonzentrationsZeit-Diagramm:
5.3 Präklinische Daten zur Sicherheit
Es liegen Untersuchungen zur subchronischen und chronischen Toxizität an verschiedenen Tierspezies vor. Ratten erhielten 6 Monate per os 100 bis 900 mg Metamizol pro kg KG. In der höchsten Dosis (900 mg pro kg KG) wurde nach 13 Wochen eine Vermehrung der Retikulozyten und der Heinz’schen Innenkörper beobachtet.
Hunde erhielten 6 Monate Metamizol in Dosen von 30 bis 600 mg pro kg KG. Dosisabhängig wurden ab 300 mg pro kg KG eine hämolytische Anämie sowie funktionelle Nieren- und Leberveränderungen beobachtet.
Für Metamizol liegen aus In-vitro- und In-vivo-Untersuchungen widersprüchliche Ergebnisse in den gleichen Testsystemen vor.
In Langzeituntersuchungen an Ratten ergaben sich keine Hinweise auf ein tumorerzeugendes Potenzial. In zwei von drei Langzeituntersuchungen an der Maus wurden in hohen Dosen vermehrt Leberzelladenome beobachtet.
Embryotoxizitätsstudien an Ratten und Kaninchen haben keine Hinweise auf teratogene Wirkungen ergeben.
Embryoletale Effekte wurden bei Kaninchen ab einer noch nicht maternaltoxischen täglichen Dosis von 100 mg pro kg KG beobachtet. Bei Ratten traten embryoletale Wirkungen bei Dosen im maternaltoxischen Bereich auf. Tägliche Dosen oberhalb von 100 mg pro kg KG führten bei Ratten zu einer Verlängerung der Tragzeit und zu einer Beeinträchtigung des Geburtsvorgangs mit erhöhter Sterblichkeit von Mutter- und Jungtieren.
Fertilitätsprüfungen zeigten eine leicht verringerte Trächtigkeitsrate bei der Elterngeneration bei einer Dosis oberhalb von 250 mg pro kg KG und Tag. Die Fertilität der F1-Generation wurde nicht beeinträchtigt.
Die Metaboliten von Metamizol gehen in die Muttermilch über. Es liegen keine Erfahrungen über deren Auswirkungen auf den Säugling vor.
6. PHARMAZEUTISCHE ANGABEN
6.1 Liste der sonstigen Bestandteile
Wasser für Injektionszwecke.
6.2 Inkompatibilitäten
Wegen der Möglichkeit von Inkompatibilitäten wird empfohlen, die Injektionslösung nicht mit anderen Therapeutika gemischt zu injizieren oder zu infundieren (zur Mischbarkeit mit Infusionslösungen siehe auch Abschnitt 4.2).
6.3 Dauer der Haltbarkeit
5 Jahre.
6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
In der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen.
6.5 Art und Inhalt des Behältnisses
Novalgin 1 g-Injektionslösung
Braune, röhrenförmige Glasampullen, verpackt in Faltschachteln zu 5 x 2 ml, 10 x 2 ml sowie Klinikpackungen zu 96 x 2 ml und 100 x 2 ml.
Novalgin 2,5 g-Injektionslösung
Braune, röhrenförmige Glasampullen, verpackt in Faltschachteln zu 4 x 5 ml, 6 x 5 ml sowie Klinikpackungen zu 20 x 5 ml und 24 x 5 ml.
6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung
Keine besonderen Anforderungen.
7. INHABER DER ZULASSUNG
Sanofi-Aventis Deutschland GmbH 65926 Frankfurt am Main
Postanschrift:
Postfach 80 08 60 65908 Frankfurt am Main
Telefon: (01 80) 2 22 20 10* Telefax: (01 80) 2 22 20 11* E-Mail: medinfo.de@sanofi.com
8. ZULASSUNGSNUMMERN
Novalgin 1 g-Injektionslösung 6196457.00.03
Novalgin 2,5 g-Injektionslösung 6196457.01.03
9. DATUM DER ERTEILUNG DER ZULASSUNG/VERLÄNGERUNG DER ZULASSUNG
Datum der Erteilung der Zulassung: 31.03.1998
Datum der letzten Verlängerung der Zulassung: 07.08.2006
10. STAND DER INFORMATION
Mai 2014
11. VERKAUFSABGRENZUNG
V erschreibungspflichtig.
0,06 €/Anruf (dt. Festnetz); max. 0,42 €/min (Mobilfunk).
13