Octanate 500
Fachinformation
1. BEZEICHNUNG DES ARZNEIMITTELS
OCTANATE 250 / 500 / 1000
Pulver und Lösungsmittel zur Herstellung einer Injektionslösung
2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
Octanate ist als Pulver und Lösungsmittel (Wasser für Injektionszwecke) zur Herstellung einer Injektionslösung erhältlich und enthält pro Durchstechflasche 250 I.E., 500 I.E. bzw. 1000 I.E. Blutgerinnungsfaktor VIII vom Menschen.
Das rekonstituierte Präparat enthält ca. 50 I.E./ml (Octanate 250), 100 I.E./ml (Octanate 500) bzw. 200 I.E./ml (Octanate 1000) Blutgerinnungsfaktor VIII, wenn es in 5 ml Wasser für Injektionszwecke (Ph. Eur.) aufgelöst worden ist.
Die Bestimmung der Aktivität (I.E.) erfolgt mit der chromogenen Methode gemäß Europäischem Arzneibuch. Die spezifische Aktivität von Octanate 250/500/1000 beträgt ca. 100 I.E./mg Protein.
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
3. DARREICHUNGSFORM
Pulver und Lösungsmittel zur Herstellung einer Injektionslösung.
4. KLINISCHE ANGABEN
4.1 Anwendungsgebiete
Prophylaxe (vorbeugende Dauerbehandlung) und Therapie von Blutungen bei
- Hämophilie A (angeborener Faktor-VIII-Mangel),
- Allen Formen von erworbenem Faktor-VIII-Mangel,
- Hemmkörperhämophilie mit Faktor-VIII-Inhibitor
Octanate enthält keinen von Willebrand-Faktor in pharmazeutisch wirksamer Menge und ist daher nicht für die Behandlung des von Willebrand-Syndroms indiziert.
4.2 Dosierung und Art der Anwendung
Die Behandlung sollte anfänglich unter Überwachung eines Arztes erfolgen, der mit der Therapie der Hämophilie A vertraut ist.
Dosierung
Dosierung und Dauer der Therapie hängen vom Schweregrad des Faktor-VIII-Mangels, Ort und Ausmaß der Blutung und dem klinischen Zustand des Patienten ab.
Die Menge der verabreichten Faktor-VIII-Einheiten wird in Internationalen Einheiten (LE.) angegeben, bezogen auf den aktuellen WHO Standard für Faktor-VIII-Präparate. Die Faktor-VIII-Aktivität im Plasma wird entweder als Prozentsatz (relativ zu normalem menschlichem Plasma) oder in Internationalen Einheiten (relativ zum Internationalen Standard für Faktor VIII im Plasma) angegeben.
Eine Faktor-VIII-Einheit entspricht der Menge Faktor VIII, die sich in 1 ml humanem Normalplasma befindet. Die Dosierungsberechnung basiert auf den Ergebnissen von klinischen Studien mit Octanate: Die Gabe einer Einheit (I.E.) Faktor VIII pro kg Körpergewicht erhöht den Faktor-VIII-Spiegel im Mittel um ca. 2 %.
Die erforderliche Dosis wird gemäß der folgenden Formel berechnet:
Körpergewicht (kg) x gewünschter Faktor-VIII-Anstieg (%) [I.E./dl] x 0,5 = Anzahl Einheiten (Dosis)
Beispiel für einen 80 kg schweren Patienten:
Gewünschter Anstieg der Faktor-VIII-Aktivität um 50%
Erforderliche Dosis in Einheiten:
80 kg x 50 % x 0,5 I.E. = 2.000 I.E.
Die Dosierung und Häufigkeit der Anwendung sollte stets der klinischen Wirksamkeit im Einzelfall angepasst werden.
Bei den folgenden Blutungsereignissen soll die Faktor-VIII-Aktivität (in % der Norm) während des entsprechenden Zeitraums nicht unter den angegebenen Wert abfallen. Die folgende Tabelle dient als Empfehlung für die Dosierung bei Blutungsereignissen und chirurgischen Eingriffen:
|
Schwere der Blutung/Art |
Benötigter Faktor-VIII- |
Häufigkeit der Dosierung |
|
des chirurgischen Eingriffs |
Plasma-Spiegel (%) |
(Stunden)/Behandlungsdauer (Tage) |
|
Blutungen | ||
|
Gelenkblutungen im Frühstadium, |
20 - 40 |
Injektion alle 12 bis 24 Stunden; |
|
Muskelblutungen, Blutungen im |
mindestens 1 Tag, bis die Blutung | |
|
Mundbereich |
sistiert bzw. Wundheilung erreicht ist. | |
|
Ausgeprägtere Gelenkblutungen, |
30 - 60 |
Injektion alle 12 bis 24 Stunden für 3 bis |
|
Muskelblutungen oder Hämatome |
4 Tage oder länger wiederholen, bis die Schmerzen und Beeinträchtigungen beseitigt sind. | |
|
Lebensbedrohliche Blutungen |
60 - 100 |
Injektion alle 8 bis 24 Stunden wiederholen, bis die Bedrohung vorüber ist. |
|
Chirurgische Eingriffe | ||
|
Kleinere Eingriffe einschließlich |
30 - 60 |
Injektion alle 24 Stunden, bis die |
|
Zahnextraktionen |
Wundheilung erreicht ist. | |
|
Größere Eingriffe |
80 - 100 |
Injektion alle 8 bis 24 Stunden wie- |
|
(prä- und post-operativ) |
derholen, bis ausreichende Wundheilung erreicht ist; dann für mindestens weitere 7 Tage einen Faktor-VIII-Spiegel von 30 % bis 60 % aufrechterhalten. |
Um Anstieg und Aufrechterhaltung des Faktor-Vlll-Spiegels zu kontrollieren, ist eine gerinnungsanalytische Überwachung dringend zu empfehlen. Dies gilt besonders für die Erhaltungsdosis nach erfolgtem chirurgischen Eingriff bis zum Abschluss der Wundheilung. Chirurgische Eingriffe erfordern die in der Tabelle angegebenen Faktor-VIII-Plasmaspiegel.
Zur Langzeitprophylaxe werden bei schwerer Hämophilie A 20 - 40 I.E. Octanate pro kg Körpergewicht im Abstand von 2-3 Tagen verabreicht. Auch hier ist eine individuelle Anpassung der Dosierung je nach klinischer Situation erforderlich.
In einer klinischen Prüfung mit 15 Kindern unter 6 Jahren wurde festgestellt, dass die Dosierungsempfehlungen auch für Kinder unter 6 Jahren übernommen werden können.
Die Patienten sollen auf die Entwicklung von Hemmkörpern gegen Faktor VIII überwacht werden. Wenn die erwarteten Spiegel der Faktor-VIII-Aktivität im Plasma nicht erreicht werden, oder wenn die Blutung nicht mit einer entsprechenden Dosis beherrscht wird, sollte ein Test zum Nachweis von Faktor-VIII-Hemmkörpern durchgeführt werden. Bei Patienten mit hohen Hemmkörperspiegeln kann die Faktor-VIII-Behandlung unwirksam sein und es sollten andere Behandlungsmöglichkeiten erwogen werden.
Die Betreuung dieser Patienten sollte nur durch Ärzte mit ausreichender Erfahrung in der Behandlung von Hämophiliepatienten erfolgen.
Siehe auch Abschnitt 4.4.
Hinweise zur Rekonstitution des Arzneimittels vor der Anwendung, siehe Abschnitt 6.6. Die Lösung wird langsam intravenös infundiert. Die empfohlene Injektionsgeschwindigkeit beträgt 2-3 ml pro Minute.
4.3 Gegenanzeigen
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Wie bei jedem intravenös zu verabreichenden Proteinpräparat sind allergische Reaktionen möglich. Das Präparat enthält außer Faktor VIII noch Spuren anderer humaner Proteine. Die Patienten sollten über frühe Anzeichen allergischer Reaktionen informiert werden wie z. B. Quaddeln, generalisierte Urtikaria, Engegefühl in der Brust, keuchende Atmung, Hypotonie und Anaphylaxie. Falls diese Symptome nach der Behandlung auftreten, sollte der Patient die Anwendung des Präparates sofort unterbrechen und seinen Arzt informieren.
Bei einem Schock sind die aktuellen medizinischen Richtlinien zur Schockbehandlung zu beachten.
Zu den Standardmaßnahmen zur Verhinderung von Infektionen infolge der Verwendung von aus menschlichem Blut oder Plasma hergestellten Medikamenten zählen die Auswahl der Spender, Untersuchung der einzelnen Spenden und Plasmapools auf spezifische Infektionsmarker sowie wirksame Produktionsschritte zur Inaktivierung/Entfernung von Viren. Dennoch kann bei der Verabreichung von Medikamenten, die aus menschlichem Blut oder Plasma hergestellt wurden, die Möglichkeit von Infektionskrankheiten durch die Übertragung von Infektionserregern nicht völlig ausgeschlossen werden. Dies gilt auch für bisher unbekannte oder neu auftretende Viren und andere Krankheitserreger. Die ergriffenen Maßnahmen gelten als wirksam gegen umhüllte Viren wie HIV, HBV und HCV und gegen das nicht-umhüllte Virus HAV. Die Maßnahmen können bei nicht-umhüllten Viren wie Parvovirus B19 von begrenzter Wirksamkeit sein. Parvovirus-B19-Infektionen können schwerwiegende Folgen für schwangere Frauen (fetale Infektion) und für Personen mit Immunmangelkrankheiten oder gesteigerter Erythropoese (z.B. hämolytische Anämie) haben.
Bei Patienten, die regelmäßig/wiederholt Präparate aus menschlichem Plasma erhalten, wird grundsätzlich eine Impfung gegen Hepatitis A und B empfohlen.
Die Bildung neutralisierender Antikörper (Hemmkörper) gegen Faktor VIII ist eine bekannte Komplikation bei der Behandlung von Hämophilie-A-Patienten. Diese Hemmkörper sind gewöhnlich IgG Immunglobuline, die sich gegen die Faktor-VIII-Gerinnungsaktivität richten. Sie werden mittels modifiziertem Test in Bethesda-Einheiten (BE) pro ml Plasma quantifiziert. Das Risiko der Bildung von Hemmkörpern korreliert mit der Anzahl der Expositionstage mit Faktor VIII, wobei das Risiko in den ersten 20 Expositionstagen am höchsten ist. In seltenen Fällen entwickeln sich Hemmkörper nach mehr als 100 Expositionstagen. Patienten, die mit Gerinnungsfaktor VIII vom Menschen behandelt wurden, sollen durch geeignete klinische Beobachtung und Labortests sorgfältig bezüglich der Entwicklung von Hemmkörpern beobachtet werden. Siehe auch Abschnitt 4.8.
Octanate 250 enthält weniger als 1 mmol (23 mg) Natrium pro Flasche.
Octanate 500/1000 enthält bis zu 1,75 mmol (40 mg) Natrium pro Flasche. Dies ist bei Patienten zu berücksichtigen, die auf eine natriumarme Ernährung achten müssen.
Es wird auf die Dokumentationspflicht gemäß Transfusionsgesetz hingewiesen.
4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Wechselwirkungen mit anderen Medikamenten sind bisher nicht bekannt geworden.
4.6 Fertilität, Schwangerschaft und Stillzeit
Reproduktionsstudien am Tier wurden mit Faktor VIII nicht durchgeführt.
Aufgrund des seltenen Vorkommens der Hämophilie A bei Frauen liegen keine Erfahrungen über die Anwendung von Faktor VIII während der Schwangerschaft und Stillzeit vor. Daher sollte Faktor VIII in der Schwangerschaft und Stillzeit nur bei klarer Indikationsstellung angewendet werden.
4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Es gibt keine Hinweise darauf, dass Octanate die Fähigkeit zur aktiven Teilnahme am Straßenverkehr oder zum Bedienen von Maschinen beeinträchtigt.
4.8 Nebenwirkungen
Die Häufigkeit der einzelnen Nebenwirkungen wurde anhand folgender Kriterien bestimmt:
Sehr häufig (> 1/10)
Häufig (> 1/100 bis < 1/10)
Gelegentlich (> 1/1.000 bis < 1/100)
Selten (> 1/10.000 bis < 1/1.000)
Sehr selten (<1/10.000).
Überempfindlichkeitsreaktionen oder allergische Reaktionen (die auch Angioödem, Brennen und Stechen an der Infusionsstelle, Schüttelfrost, Hautrötung mit Hitzegefühl, generalisierte Nesselsucht, Kopfschmerzen, Nesselausschlag, Hypotonie, Antriebslosigkeit, Übelkeit, Unruhe, Tachykardie, Engegefühl in der Brust, Zittern, Erbrechen und Stridor mit einschließen können) wurden bei Faktor-VIII-Präparaten mit seltener Häufigkeit beobachtet und können sich in einigen Fällen zu schwerer Anaphylaxie (einschließlich Schock) entwickeln.
In seltenen Fällen wurde Fieber beobachtet.
|
Systemorganklasse |
Selten |
Sehr selten |
|
Erkrankungen des Immunsystems |
Überempfindlichkeitsreaktionen |
Anaphylaktischer Schock |
|
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort |
Fieber | |
|
Untersuchungen |
Faktor-VIII-Antikörper |
Patienten mit Hämophilie A können neutralisierende Antikörper (Hemmkörper) gegen Faktor VIII entwickeln. Wenn solche Hemmkörper auftreten, manifestiert sich der Zustand als unzureichende klinische Antwort. In solchen Fällen wird empfohlen, ein spezialisiertes Hämophiliezentrum aufzusuchen.
Die Bildung von Hemmkörpern wird vorrangig bei nicht vorbehandelten Patienten (PUPs) beobachtet. In einer klinischen Prüfung mit Octanate bei zuvor unbehandelten Patienten mit schwerer Hämophilie A waren zum Zeitpunkt einer Zwischenauswertung bei drei von 29 Patienten Hemmkörper aufgetreten. Bei allen 3 Patienten trat der Hemmkörper innerhalb der ersten 20 Expositionstage auf und lag initial zwischen 5 und 10 Bethesda-Einheiten. Nach 34 Monaten Bedarfsbehandlung war der Hemmkörper bei 1 der 3 Patienten nicht länger nachweisbar.
In klinischen Prüfungen mit Octanate bei 77 vorbehandelten Studienpatienten mit schwerer Hämophilie A trat kein Hemmkörper auf. Spontanberichte über den Verdacht der Hemmkörperbildung liegen nicht vor.
Informationen zum Infektionsrisiko siehe Abschnitt 4.4.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-RisikoVerhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel, Paul-Ehrlich-Institut, Paul-Ehrlich-Str. 51-59, 63225 Langen, Telefon: +49 6103 77 0, Telefax: +49 6103 77 1234, Website: www.pei.de anzuzeigen.
4.9 Überdosierung
Symptome einer Überdosierung mit Blutgerinnungsfaktor VIII vom Menschen wurden nicht berichtet.
PHARMAKOLOGISCHE EIGENSCHAFTEN
5.
5.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Antihämorrhagika, Gerinnungsfaktor VIII ATC-Code: B02BD02
Der Faktor VIII/von-Willebrand-Faktor (vWF)- Komplex besteht aus zwei Molekülen (Faktor VIII und von-Willebrand-Faktor) mit unterschiedlichen physiologischen Funktionen. Wird einem Hämophilie-A-Patienten Faktor VIII injiziert, so bindet dieser im Blutkreislauf an den von-Willebrand-Faktor. Der aktivierte Faktor VIII wirkt als Kofaktor für den aktivierten Faktor IX und beschleunigt die Bildung von aktiviertem Faktor X (Faktor Xa) aus Faktor X. Aktivierter Faktor X wandelt Prothrombin in Thrombin um. Dieses setzt dann aus Fibrinogen Fibrin frei und die Gerinnselbildung kann erfolgen.
Hämophilie A ist eine X-chromosomal-gebundene, erbliche Blutgerinnungsstörung, die sich in Form eines erniedrigten Faktor-VIII-Plasmaspiegels manifestiert. Dies führt entweder spontan oder in Folge unfallbedingter oder chirurgischer Traumata zu starken Blutungen in Gelenken, Muskeln oder inneren Organen. Durch die Substitutionstherapie werden die Faktor-VIII-Plasmaspiegel erhöht, wodurch eine temporäre Beseitigung des Faktor-VIII-Mangels ermöglicht und die Blutungsneigung vermindert wird.
5.2 Pharmakokinetische Eigenschaften
Aus humanem Plasma hergestellter Faktor VIII ist ein normaler Bestandteil des menschlichen Plasmas und verhält sich wie der körpereigene Faktor VIII. Nach Injektion verbleiben zwei Drittel bis drei Viertel des Faktor VIIIs im Kreislauf. Der erreichte Faktor-VIII-Spiegel sollte zwischen 80 und 120 % der erwarteten Faktor-VIII-Aktivität betragen.
Nach intravenöser Anwendung nimmt die Faktor-VIII-Aktivität exponentiell in zwei Phasen ab. In der Initialphase vollzieht sich die Verteilung zwischen dem intravaskulären Raum und den übrigen Verteilungsräumen (Körperflüssigkeiten) mit einer Plasma-Halbwertszeit von 3 bis 6 Stunden. In der nachfolgenden Phase liegt die Halbwertszeit zwischen 8 und 20 Stunden, mit einem Mittelwert von ca. 12 Stunden. Dies entspricht der tatsächlichen biologischen Halbwertszeit.
Die folgenden Ergebnisse wurden im Rahmen von zwei Pharmakokinetikstudien mit 10 bzw. 14 Hämophilie-A-Patienten ermittelt:
|
Wiederfindungsrate (Recovery) % x I.E.-1 x kg |
AUC*1 norm (% x h x I.E.-1 x kg) |
Halbwerts zeit (Half-life) (h) |
Mittlere Verweildauer (MRT*2) (h) |
Clearance (ml x h-1 x kg) | |
|
Studie 1, n = 10 MW± SD*3 |
2,4 ± 0,36 |
45,5 ± 17,2 |
14,3 ± 4,01 |
19,6 ± 6,05 |
2,6 ± 1,21 |
|
Studie 2, n = 14 MW ± SD*3 |
2,4 ± 0,25 |
33,4 ± 8,50 |
12,6 ± 3,03 |
16,6 ± 3,73 |
3,2 ± 0,88 |
AUC* = Fläche unterhalb der Kurve (area under the curve),
MRT*2 = mean residence time
MW± SD* = Mittelwert ± Standardabweichung (standard deviation)
5.3 Präklinische Daten zur Sicherheit
Faktor VIII (im Konzentrat) ist ein normaler Bestandteil des menschlichen Plasmas und verhält sich wie körpereigener Faktor VIII.
Die einmalige Verabreichung des Mehrfachen der bei Menschen angewendeten Dosis, bezogen auf das Kilogramm Körpergewicht, zeigt bei Labortieren keine toxische Wirkung. Toxizitätstests mit wiederholter Verabreichung sind im Tierversuch auf Grund der Antikörperbildung gegen heterologe Proteine nicht sinnvoll.
6. PHARMAZEUTISCHE ANGABEN
6.1 Liste der sonstigen Bestandteile
Natriumcitrat
Calciumchlorid
Natriumchlorid
Glycin
Wasser für Injektionszwecke
6.2 Inkompatibilitäten
Da keine Kompatibilitätsstudien durchgeführt wurden, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Es sollte nur das beigefügte Infusionsset verwendet werden, da ein Therapieversagen auf Grund der Adsorption von Faktor VIII an den Innenflächen einiger anderer Injektionssets auftreten kann.
6.3 Dauer der Haltbarkeit
Pulver und Lösungsmittel (Wasser für Injektionszwecke):
2 Jahre
Rekonstituierte Lösung:
Die rekonstituierte Lösung muss sofort verwendet werden.
6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Nicht über 25°C lagern. Das Behältnis im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen. Nicht einfrieren.
6.5 Art und Inhalt des Behältnisses
1 Packung Octanate enthält:
1 Durchstechflasche mit Pulver
1 Durchstechflasche mit 5 ml Lösungsmittel (Wasser für Injektionszwecke)
1 Gerätesatz (1 Transferset [Mix2Vial®], 1 Flügelkanüle, 1 Einmalspritze)
2 Alkoholtupfer
6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung
Bitte lesen Sie alle Anweisungen durch und befolgen Sie sie sorgfältig. Der Lösungsvorgang des Präparates und die Injektion müssen unter aseptischen Bedingungen erfolgen.
Anleitung für das Auflösen des Faktor-VIII-Konzentrates:
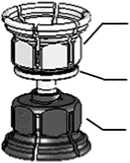
Konzentrat
adapter
(transparent)
integrierter
Filter
Lösungsmittel
adapter
(blau)
Abb. 1
1. Lösungsmittel (Wasser für Injektionszwecke) und Konzentrat in den ungeöffneten Flaschen auf Zimmertemperatur bringen, falls sie gekühlt gelagert wurden. Nicht direkt aus dem Kühlschrank verwenden.
2. Die Schutzkappen von der Konzentrat- und Lösungsmittelflasche entfernen und die Gummistopfen beider Flaschen mit einem Alkoholtupfer desinfizieren.
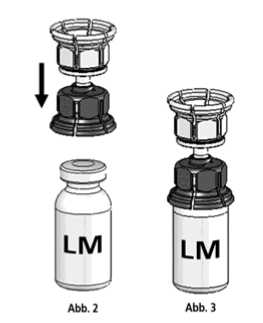
3. Die Lösungsmittelflasche auf eine feste, ebene Unterlage (z.B. Tisch) stellen. Das in Abb. 1 beschriebene Mix2Vial-Set mit dem blauen Adapter auf die Lösungsmittelflasche (LM) aufsetzen und bis zum Anschlag nach unten drücken (Abb. 2+3).
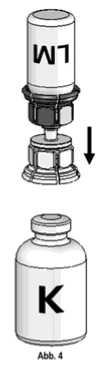
4. Die Konzentratflasche (K) auf eine feste, ebene Unterlage stellen. Die Lösungsmittelflasche (LM) mit dem Mix2Vial-Set umdrehen und senkrecht mit dem transparenten Ende auf die Konzentratflasche (K) aufsetzen und bis zum Anschlag nach unten drücken (Abb. 4). Das Vakuum in der Konzentratflasche saugt das Wasser an.
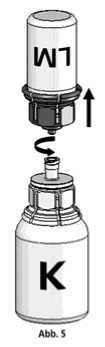
5. Die Konzentratflasche mit Mix2Vial-Set und der verbundenen Lösungsmittelflasche leicht schwenken (nicht schütteln) bis das Produkt vollständig gelöst ist. Das Konzentrat löst sich bei Zimmertemperatur spätestens nach 10 Minuten vollständig. Dabei ist eine leichte Schaumbildung möglich, die sich auflösen wird. Die Lösungsmittelflasche zusammen mit dem blauen Adapter von der Konzentratflasche abdrehen (Abb. 5) und die Lösungsmittelflasche mit dem blauen Adapter verwerfen.
Nach Auflösen in dem beigefügten Wasser für Injektionszwecke wird Octanate intravenös verabreicht. Die Lösung sollte klar und farblos oder leicht opaleszent sein. Verwenden Sie keine Lösungen, die trüb sind oder Ablagerungen aufweisen. Die rekonstituierte Lösung muss sofort verwendet werden.
Injektion:
Der Puls sollte vor und während der Injektion gemessen werden. Eine deutliche Erhöhung der Pulsfrequenz klingt normalerweise nach Verlangsamen oder Unterbrechen der Injektion schnell wieder ab.
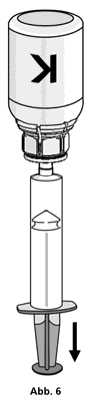
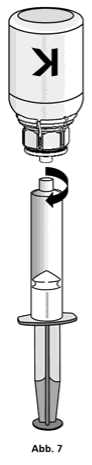
1. Die Spritze mit dem transparenten Adapter der Konzentratflasche verbinden. Die Flasche samt Einmalspritze umdrehen und das aufgelöste Präparat in die Spritze aufziehen (Abb. 6). Die Injektionslösung in der Spritze sollte klar oder leicht schillernd sein. Nachdem das Produkt in die Spritze überführt wurde, den Spritzenzylinder fassen und die Spritze vom transparenten Adapter der Konzentratflasche entfernen (Abb. 7). Verwerfen Sie die Konzentratflasche mit dem Adapter.
2. Vorgesehene Injektionsstelle mit einem Alkoholtupfer desinfizieren.
3. Die beigepackte Flügelkanüle auf die Spritze aufsetzen.
4. Stechen Sie die Flügelkanüle in die gewählte Vene. Wenn Sie die Vene vor der Punktion gestaut haben, damit Sie sie besser sehen können, müssen Sie die Stauung öffnen, bevor Sie mit der Injektion beginnen. Es darf kein Blut in die Spritze gelangen, da dies zur Bildung von Blutgerinnseln führen könnte.
5. Injizieren Sie die Lösung langsam in die Vene, wobei die Injektionsgeschwindigkeit höchstens 2 - 3 ml pro Minute betragen sollte
Wenn Sie mehr als eine Flasche des Konzentrates benötigen, kann die Flügelkanüle in der Vene belassen werden. Zum Herstellen der gebrauchsfertigen Lösung immer ein neues Mix2Vial-Set benutzen
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
7. INHABER DER ZULASSUNG
OCTAPHARMA GmbH Elisabeth-Selbert-Str. 11 40764 Langenfeld E-Mail: info@octapharma.de www.octapharma.de
8. ZULASSUNGSNUMMER(N) 10500a/97-1 (OCTANATE 250)
10500a/97-2 (OCTANATE 500)
10500a/97-3 (OCTANATE 1000)
9. DATUM DER ERTEILUNG DER ZULASSUNG/VERLÄNGERUNG DER ZULASSUNG
04.08.1998
10. STAND DER INFORMATION
April 2015
11. HERKUNFTSLAND DES BLUTPLASMAS
Deutschland, Estland, Finnland, Kroatien, Luxemburg, Norwegen, Österreich, Portugal, Schweden, Schweiz, Slowenien, Spanien, Tschechische Republik, Ungarn, USA
12. VERSCHREIBUNGSSTATUS
Verschreibungspflichtig
12/11