Palladon Retard 16Mg
Fachinformation (SPC)
1. Bezeichnung der Arzneimittel
Palladon retard 4 mg Palladon® retard 8 mg Palladon® retard 16 mg Palladon® retard 24 mg
2. Qualitative und quantitative Zusammensetzung
Wirkstoff: Hydromorphonhydrochlorid Je 1 Palladon retard Hartkapsel enthält
4 mg Hydromorphonhydrochlorid (entsprechend 3,56 mg Hydromorphon) bzw.
8 mg Hydromorphonhydrochlorid (entsprechend 7,12 mg Hydromorphon), bzw.
16 mg Hydromorphonhydrochlorid (entsprechend 14,24 mg Hydromor-phon), bzw.
24 mg Hydromorphonhydrochlorid (entsprechend 21,36 mg Hydromor-phon).
Die vollständige Auflistung der sonstigen Bestandteile siehe Abschnitt 6.1.
3. Darreichungsform
Hartkapsel, retardiert („Retardkapsel")
Hartkapsel mit durchsichtigem Unterteil und farbigem Oberteil, die weiße bis cremefarbene, sphärische Retard-Pellets enthält:
4 mg: hellblau, mit der Aufschrift „HCR 4",
8 mg: rosa, mit der Aufschrift „HCR 8",
16 mg: hellbraun, mit der Aufschrift „HCR 16",
24 mg: dunkelblau, mit der Aufschrift „HCR 24".
4. Klinische Angaben
4.1. Anwendungsgebiete Behandlung von starken Schmerzen.
4.2. Dosierung, Art und Dauer der Anwendung Art der Anwendung
Zum Einnehmen
Die Hartkapseln sind mit ausreichend Flüssigkeit unzerkaut einzunehmen.
Die Dosierung von Palladon retard muss der Stärke der Schmerzen und der individuellen Reaktion des Patienten angepasst werden.
Dabei sollte ein Zeitintervall von 12 Stunden nicht unterschritten werden. Bei der Behandlung chronischer Schmerzen ist der Dosierung nach festem Zeitplan der Vorzug zu geben.
Die Dosis sollte schrittweise bis zur optimalen Schmerzstillung gesteigert werden.
Grundsätzlich sollte eine ausreichend hohe Dosis gegeben werden und gleichzeitig die im Einzelfall kleinste analgetisch wirksame Dosis angestrebt werden.
Wie bei jedem anderen starken Opioid sollte eine angemessene Prophylaxe bekannter Nebenwirkungen (zum Beispiel Obstipation) in Betracht gezogen werden.
Dauer der Anwendung
Palladon retard sollte nicht länger als unbedingt notwendig verabreicht werden. Wenn entsprechend Art und Schwere der Erkrankung eine Langzeitbehandlung erforderlich ist, sollte eine sorgfältige und regelmäßige Überprüfung sicherstellen, ob und in welchem Ausmaß eine Weiterbehandlung notwendig ist.
Therapieende
Bei Patienten mit einer physischen Abhängigkeit von Opioiden kann ein abruptes Absetzen von Hydromorphon zu einem Abstinenz-/Entzugssyndrom führen. Ist eine Beendigung der Therapie mit Hydromorphon indiziert, sollte die Hydromorphon-Dosis deshalb alle 2 Tage um jeweils 50% verringert werden, bis die niedrigstmögli-che Dosis erreicht ist, bei der die Therapie sicher beendet werden kann. Falls Entzugserscheinungen auftreten, ist die Dosisreduktion abzubrechen. Die Dosis sollte dann geringfügig erhöht werden, bis die Anzeichen und Symptome eines Opioidentzugs verschwinden. Danach ist die Dosisreduktion von Hydromorphon fortzusetzen, jedoch mit längeren Zeitintervallen zwischen jeder Hydromorphon-Dosisreduktion oder indem die Reduktion mit einer äquianalgetischen Dosis eines anderen Opioids fortgeführt wird.
Erwachsene und Jugendliche ab 12 Jahre:
Die Anfangsdosis von Palladon retard beträgt im Allgemeinen 4 mg alle 12 Stunden. Die Dosis kann abhängig von der Schmerzlinderung vorsichtig titriert werden.
Bei Patienten, die regelmäßig mit Opioiden behandelt werden, kann eine höhere Anfangsdosierung von Palladon® retard in Abhängigkeit der vorherigen OpioidTagesdosis notwendig sein.
Kinder
Palladon retard wird für die Anwendung bei Kindern unter 12 Jahren nicht empfohlen, da bei Kindern unter 12 Jahren keine ausreichend dokumentierten Erfahrungen vorliegen.
Ältere Patienten
Bei älteren Patienten kann eventuell mit einer geringeren Dosierung als bei anderen Erwachsenen eine ausreichende Analgesie erzielt werden.
Patienten mit Leber und/oder Nierenfunktionsstörungen
Diese Patienten benötigen möglicherweise niedrigere Dosen als andere Patientengruppen, um eine ausreichende Analgesie zu erreichen. Sie sollen vorsichtig entsprechend der Wirkung eingestellt werden (siehe Abschnitt 5.2).
4.3. Gegenanzeigen
Hydromorphon ist kontraindiziert bei
- Überempfindlichkeit gegenüber Hydromorphon oder einem der sonstigen Bestandteile
- Atemdepression
- schwerer chronisch obstruktiver Atemwegserkrankung
- Koma
- akutem Abdomen
- paralytischem Ileus
- gleichzeitiger Gabe von Monoaminoxidasehemmern oder wenn diese innerhalb der letzten 14 Tage abgesetzt wurden.
4.4. Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Eine Atemdepression ist die bedeutsamste Gefährdung einer Opioidüberdosierung. Vorsicht bei der Anwendung ist geboten bei opioidabhängigen Patienten, bei Patienten mit Kopfverletzungen (wegen des Risikos eines erhöhten Gehirndrucks), Krampfleiden, Alkoholismus, Delirium tremens, toxischer Psychose, Hypotonie bei Hy-povolämie, Bewusstseinsstörungen, Gallenwegserkrankungen, Gallen- oder Nierenkolik, Pankreatitis, obstruktive oder entzündliche Darmerkrankungen, Prostatahypertrophie, Nebennierenrindeninsuffizienz (z.B. Morbus Addison), Hypothyreose, chronisch obstruktiver Atemwegserkrankung, verminderter Atemreserve, bei älteren oder geschwächten Patienten und bei Patienten mit schwerer Beeinträchtigung der Nieren- oder Leberfunktion (siehe Abschnitt 4.2). Bei allen vorgenannten Patienten kann eine niedrigere Dosierung ratsam sein.
®
Bei längerfristiger Anwendung von Palladon retard kann es zur Entwicklung einer Toleranz mit der Erfordernis höherer Dosen zum Erzielen des erwünschten analgetischen Effektes kommen. Eine Kreuztoleranz zu anderen Opioiden kann bestehen. Die chronische Anwendung von Palladon® retard kann zu physischer Abhängigkeit führen, und bei abrupter Beendigung der Therapie kann ein Entzugsyndrom auftreten. Wenn die Therapie mit Hydromorphon nicht mehr länger erforderlich ist, kann es ratsam sein, die Tagesdosis allmählich zu reduzieren, um das Auftreten der Symptome eines Entzugssyndroms zu vermeiden.
Hydromorphon besitzt ähnlich wie andere starke Opioide ein Missbrauchspotenzial. Hydromorphon kann daher von Personen mit latenten oder manifesten Suchterkrankungen bewusst missbraucht werden. Eine psychische Abhängigkeit (Arzneimittelsucht) kann sich nach Gabe opioidhaltiger Analgetika wie Palladon® retard entwickeln. Daher ist Palladon® retard bei anamnestischem Alkohol- oder Arzneimittelmissbrauch nur mit besonderer Vorsicht zu verordnen.
Um die Retardierung der in den Kapseln enthaltenen Pellets nicht zu beeinträchtigen, dürfen diese nicht zerteilt, zerkaut oder zerrieben werden. Die Anwendung zerteilter, zerkauter oder zerriebener Pellets führt zu einer schnellen Freisetzung und zur Resorption einer möglicherweise letalen Dosis von Hydromorphon (siehe Abschnitt 4.9).
Bei gleichzeitiger Einnahme von Alkohol und Palladon retard können vermehrt Nebenwirkungen von Palladon® retard auftreten. Die gleichzeitige Einnahme sollte vermieden werden.
Palladon retard ist nur für die orale Einnahme bestimmt. Eine missbräuchliche parenterale Verabreichung von Palladon® retard kann zu schwerwiegenden, potentiell letalen unerwünschten Ereignissen führen.
Palladon retard sollte nicht eingesetzt werden, wenn die Möglichkeit besteht, dass ein paralytischer Ileus auftritt. Sollte ein paralytischer Ileus vermutet werden oder während der Behandlung auftreten, muss die Behandlung mit Hydromorphon sofort abgebrochen werden.
Bei einer Hyperalgesie, die sehr selten insbesondere bei hoher Dosierung auftreten kann, wird eine weitere Dosiserhöhung von Palladon® retard zu keiner weiteren
Schmerzreduktion führen. Eine Dosisreduktion oder der Wechsel zu einem anderen Opioid kann dann erforderlich werden.
Palladon retard wird präoperativ und in den ersten 24 Stunden postoperativ wegen des gegenüber Nichtoperierten in der postoperativen Phase höheren Risikos eines Ileus nicht empfohlen. Danach sollte Palladon® retard - insbesondere bei abdominalen Eingriffen - mit Vorsicht angewendet werden.
Patienten, die einer anderen zusätzlichen Schmerztherapie (z. B. Operation, Plexusblockade) unterzogen werden, sollten 12 Stunden vor dem Eingriff kein Hydromor-phon mehr erhalten. Falls eine Weiterbehandlung mit Palladon® retard indiziert ist, sollte die Dosierung nach dem Eingriff den neuen Erfordernissen entsprechend eingestellt werden.
Es ist zu beachten, dass Patienten nach erfolgter Einstellung (Titration) auf wirksame Dosen eines bestimmten Opioides nicht ohne ärztliche Beurteilung und sorgfältige bedarfsorientierte Neueinstellung auf ein anderes Opioid umgestellt werden sollten. Andernfalls ist eine kontinuierliche, analgetische Wirkung nicht gewährleistet.
Palladon retard 8 mg, 16 mg, 24 mg sind nicht für eine initiale Opioid-Therapie geeignet. Die höheren Wirkstärken von Palladon® retard (8 mg, 16 mg, 24 mg) dürfen nur bei Patienten angewendet werden, bei denen im Rahmen einer langfristigen Schmerzbehandlung mit niedriger dosierten Hydromorphon-Präparaten (Palladon® retard 4 mg) oder anderen vergleichbar starken Schmerzmitteln keine ausreichende Schmerzfreiheit mehr erreicht werden kann.
Bei bestehender Nebennierenrinden-Insuffizienz sollte die Plasmakortisolkonzentration kontrolliert und ggf. Kortikoide zugeführt werden.
Die Anwendung von Palladon retard kann bei Dopingkontrollen zu positiven Ergebnissen führen.
4.5. Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Die gleichzeitige Anwendung von zentralwirksamen Arzneimitteln wie Tranquilizer, Anästhetika wie z. B. Barbiturate, Hypnotika und Sedativa, Neuroleptika, Antidepressiva, Antihistaminika/Antiemetika und andere Opioide kann zu einer zusätzlichen dämpfenden Wirkung auf das Zentralnervensystem führen. Hierdurch können z. B. Sedierung und Atemdepression eintreten.
Alkohol kann die pharmakodynamischen Effekte von Palladon retard verstärken. Die gleichzeitige Einnahme sollte vermieden werden.
Monoaminoxidase-Hemmer (MAO-Hemmer) können bei gleichzeitiger Anwendung mit Opioiden entweder stimulierend oder hemmend auf das ZNS wirken oder zu einer Hypotonie oder Hypertonie führen. Palladon retard ist bei gleichzeitiger Therapie mit MAO-Hemmern kontraindiziert (siehe Abschnitt 4.3).
Ebenso wie andere Opioide kann Palladon retard die neuromuskuläre Blockadewirkung von Muskelrelaxantien erhöhen und zu einer verstärkten Atemdepression führen.
Es wurden keine Wechselwirkungsstudien durchgeführt.
4.6. Schwangerschaft und Stillzeit
®
Die Anwendung von Palladon retard während der Schwangerschaft und Stillzeit wird nicht empfohlen.
Schwangerschaft
Für Hydromorphon liegen keine klinischen Daten über exponierte Schwangere vor.
Palladon retard sollten während der Schwangerschaft und während der Geburt wegen verminderter Uteruskontraktilität und der Gefahr einer Atemdepression beim Neugeborenen nicht eingenommen werden. Eine chronische Einnahme während der Schwangerschaft kann zu Entzugserscheinungen beim Neugeborenen führen.
Tierexperimentelle Untersuchungen haben eine Reproduktionstoxizität gezeigt (siehe Abschnitt 5.3).
Stillzeit
Es sind keine Daten zur Anwendung von Palladon retard während der Stillzeit verfügbar. Palladon® retard sollte deshalb von stillenden Müttern nicht eingenommen werden; wenn die Einnahme erforderlich ist, sollte abgestillt werden.
4.7. Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Hydromorphon kann die Verkehrstüchtigkeit und die Fähigkeit, Maschinen zu bedienen, beeinträchtigen. Dies ist insbesondere zu Beginn einer Hydromorphon-Therapie, nach Dosiserhöhung oder Präparatewechsel sowie beim Zusammenwirken von Hydromorphon mit ZNS dämpfenden Substanzen zu erwarten. Bei einer stabilen Therapie sind Beschränkungen nicht zwangsläufig erforderlich. Deshalb sollten Patienten mit ihrem behandelnden Arzt besprechen, ob sie Autofahren oder Maschinen bedienen dürfen.
4.8. Nebenwirkungen
Die am häufigsten berichteten Nebenwirkungen sind Übelkeit (vor allem zu Beginn der Therapie) und Obstipation.
Bei der Bewertung von Nebenwirkungen werden folgende Häufigkeiten zugrunde gelegt:
|
Sehr häufig |
>1/10 |
|
Häufig |
>1/100 bis <1/10 |
|
Gelegentlich |
>1/1.000 bis <1/100 |
|
Selten |
>1/10.000 bis <1/1.000 |
|
Sehr selten |
<1/10.000 |
|
Nicht bekannt |
Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar |
Erkrankungen des Immunsystems:
Sehr selten: Überempfindlichkeitsreaktionen (einschließlich Schwellungen im
Bereich des Oropharynx)
Nicht bekannt: Anaphylaktische Reaktionen
Stoffwechsel- und Ernährungsstörungen:
Häufig: Appetitabnahme bis zum Appetitverlust
Psychiatrische Erkrankungen:
Häufig: Angstzustände, Verwirrtheit, Schlaflosigkeit
Depression, Dysphorie, Euphorie, Halluzinationen, Albträume
Gelegentlich:
Selten:
Abhängigkeit, Agitiertheit
Erkrankungen des Nervensystems:
Häufig: Schwindel, Somnolenz
Gelegentlich: Kopfschmerzen, Tremor, Myoklonus, Parästhesie
Selten: Krampfanfälle, Sedierung
Sehr selten: Hyperalgesie (siehe Abschnitt 4.4)
Augenerkrankungen:
Gelegentlich: Miosis, Verschwommensehen
Herzerkrankungen:
Gelegentlich: Tachykardie
Selten: Bradykardie, Palpitationen
Gefäßerkrankungen:
Häufig: Hypotonie
Erkrankungen der Atemwege, des Brustraums und des Mediastinums: Gelegentlich: Dyspnoe
Selten: Atemdepression, Bronchospasmus
Erkrankungen des Gastrointestinaltrakts:
Häufig: Obstipation, Bauchschmerzen, Mundtrockenheit, Übel
keit, Erbrechen
Gelegentlich: Dyspepsie, Diarrhoe, Geschmacksstörungen
Sehr selten: paralytischer Ileus
Leber- und Gallenerkrankungen:
Selten: Gallenkoliken, Erhöhung von Pankreasenzymen
Sehr selten: Erhöhung leberspezifischer Enzyme
Erkrankungen der Haut und des Unterhautzellgewebes: Häufig: Pruritus, Schwitzen
Gelegentlich: Hautausschlag, Urtikaria
Selten: Rötung des Gesichts
Erkrankungen der Nieren und Harnwege:
Häufig: Harnverhalten, verstärkter Harndrang
Erkrankungen der Geschlechtsorgane und der Brustdrüse: Gelegentlich: verminderte Libido, Erektionsstörungen
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort:
Häufig: Schwächezustände
Gelegentlich: Toleranz, Entzugserscheinungen*
Sehr selten: periphere Ödeme
*Entzugserscheinungen können auftreten und sich in Symptomen wie gesteigerter Erregbarkeit, Angstzuständen, Nervosität, Schlaflosigkeit, Hyperkinesie, Tremor und gastrointestinalen Symptomen äußern.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-RisikoVerhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Arzneimittel und Medizinprodukte, Abt. Pharmakovigilanz, Kurt-Georg-Kiesinger-Allee 3, D-53175 Bonn, Website: www.bfarm.de anzuzeigen.
4.9. Überdosierung
Symptome der Intoxikation:
Zeichen und Symptome einer übermäßigen Hydromorphon-Wirkung sind durch Beschwerden wie „sich komisch fühlen", schlechte Konzentrationsfähigkeit, Schläfrigkeit und möglicherweise Schwindelgefühl im Stehen gekennzeichnet. Weitere mögliche typische Symptome einer Überdosierung sind Atemdepression (Verringerung der Atemfrequenz und/oder des Atemzugvolumens, Cheyne-Stokes-Atmung, Zyanose), extreme Schläfrigkeit, Bewusstseinsstörungen bis hin zum Koma, Miosis, Erschlaffung der Skelettmuskulatur, feuchtkalte Haut, Bradykardie und Hypotension. Massive Vergiftungen können Atemstillstand, Kreislaufversagen, Herzstillstand und den Tod hervorrufen.
Therapie der Intoxikation:
Im Falle einer Überdosierung ist der kardiale und respiratorische Zustand des Patienten engmaschig zu überwachen und es sind entsprechende unterstützende Maßnahmen einzuleiten. Ein spezifischer Opioidantagonist wie Naloxon kann die Wirkungen von Hydromorphon aufheben. Es muss darauf geachtet werden, dass die Wirkungsdauer von Opioiden länger sein kann als die von Naloxon, wodurch ein Wiederauftreten der Atemdepression möglich ist. Bei Einnahme größerer Mengen Palladon® retard sollte eine Magenspülung erwogen werden.
5. Pharmakologische Eigenschaften
5.1. Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Opioid-Analgetikum; natürliches Opium-Alkaloid ATC-Code: N02A A03.
Hydromorphon ist ein g-selektiver, reiner Opioid-Agonist. Hydromorphon und verwandte Opioide wirken hauptsächlich auf das zentrale Nervensystem und den Darm.
Die Wirkungen sind vornehmlich analgetisch, anxiolytisch, antitussiv und sedativ. Darüber hinaus können Stimmungsveränderungen, Atemdepression, verminderte gastrointestinale Motilität, Übelkeit, Erbrechen und Veränderungen des endokrinen und autonomen Nervensystems auftreten.
Opioide können die Hypothalamus-Hypophysen-Nebennieren- oder -GonadenAchsen beeinflussen. Zu den Veränderungen, die beobachtet werden können, zählen ein Anstieg des Prolaktin im Serum und eine Abnahme von Kortisol und Testosteron im Plasma. Eine Manifestation klinischer Symptome aufgrund dieser Hormonveränderungen kann möglich sein.
Präklinische Studien zeigen unterschiedliche Effekte von Opioiden auf Komponenten des Immunsystems. Die klinische Bedeutung dieser Befunde ist nicht bekannt.
5.2. Pharmakokinetische Eigenschaften
Hydromorphon wird im Gastrointestinaltrakt resorbiert und unterliegt einer präsystemischen Elimination. Das Ausmaß der absoluten Bioverfügbarkeit beträgt 36,4 % (C.I. 90%: 32,7 - 40,5 %) für Palladon® retard und 32,3 % (C.I. 90%: 29,0 - 35,9 %) für die orale Hydromorphonlösung. Die relative Bioverfügbarkeit von Palladon® retard ist vergleichbar mit der Bioverfügbarkeit des normal freisetzenden Hydromor-phonhydrochlorid, jedoch mit geringerer Fluktuation der Plasmaspiegel. Die maximale Plasmakonzentration (Cmax=1,2±1,24 ng/ml) nach Einnahme von Palladon® retard wird nach 2 bis 5 Stunden (Tmax =3 (2-5) erreicht gefolgt von einer langgestreckten Plateauphase mit relativ konstantem therapeutischen Plasmaspiegel von mindestens 12 Stunden.
Plasmaspiegelverläufe (Mittelwerte)
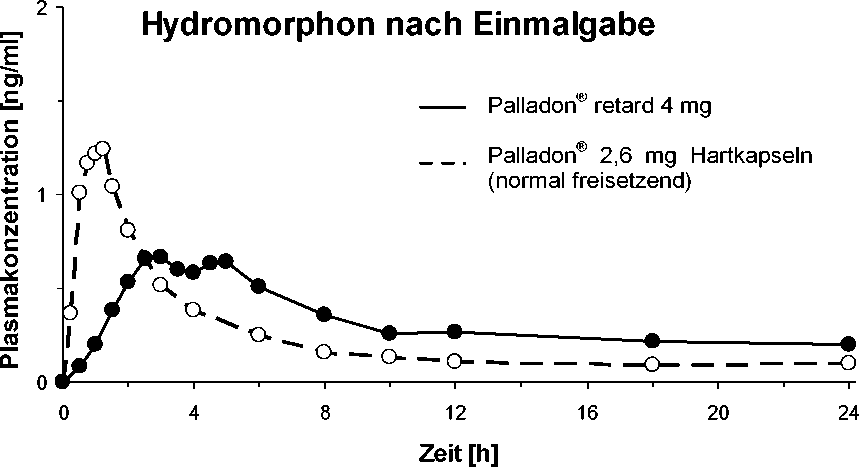
Die Plasmaproteinbindung des Hydromorphons ist gering (< 10 %), wobei dieser Prozentsatz von 2,46 ng/ml bis zu sehr hohen Plasmaspiegeln von 81,99 ng/ml, die nur bei sehr hohen Hydromorphon-Dosen erreicht werden, konstant bleibt.
Hydromorphonhydrochlorid weist ein relativ hohes Verteilungsvolumen von 1,22 ± 0,23 l/kg (C.I.: 90%: 0,97 - 1,60 l/kg, N=6 männliche Probanden) auf. Dies weist auf eine deutliche Gewebeaufnahme hin.
Aus dem Verlauf der Plasmakonzentrations-Zeit-Kurven nach einmaliger Gabe von Hydromorphonhydrochlorid 2 mg i.v. oder 4 mg oral an 6 gesunde Probanden im randomisierten Cross-over-Versuch ergab sich eine relative kurze Eliminationshalbwertszeit von 2,64 ± 0,88 Stunden (1,68-3,87 Stunden).
Hydromorphon wird durch direkte Konjugation oder durch Reduktion der Ketogruppe mit nachfolgender Konjugation metabolisiert. Nach Resorption wird Hydromorphon hauptsächlich zu Hydromorphon-3-Glukuronid, Hydromorphon-3-Glukosid und Dihydroisomorphin-6-Glukuronid metabolisiert. Zu einem kleineren Anteil wurden auch die Metabolite Dihydroisomorphin-6-Glukosid, Dihydromorphin und Dihydro-isomorphin beobachtet. Hydromorphon wird hepatisch metabolisiert und zum geringen Teil unverändert hauptsächlich renal ausgeschieden.
Hydromorphonmetaboliten wurden im Plasma, Urin und in humanen Hepatozyten-Test-Systemen festgestellt. Es gibt keine Hinweise, dass Hydromorphon in-vivo durch das Cytochrom P 450 Enzymsystem metabolisiert wird. In-vitro hemmt Hydromorphon mit einer IC50>50mM nur geringfügig die rekombinanten CYP-Isoformen, einschließlich CYP1A2, 2A6, 2C8, 2D6 und 3A4. Es ist deshalb nicht zu erwarten, dass Hydromorphon den Metabolismus von anderen Arzneistoffen, die durch diese CYP-Isoformen metabolisiert werden, inhibiert.
5.3. Präklinische Daten zur Sicherheit
An Ratten, die oral 5 mg/kg/Tag erhielten (30 mg/m /Tag, was 1,4 fach höher ist, als die für den Menschen nach Körperoberfläche errechnete, zu erwartende Dosis), wurden keine Auswirkungen auf die männliche oder weibliche Fertilität oder die Eigenschaften der Spermien beobachtet.
Hydromorphon-Dosen, welche auf das Muttertier toxisch wirkten, waren weder bei Ratten noch Kaninchen teratogen. Eine Beeinträchtigung der foetalen Entwicklung ergab sich bei Kaninchen in einer Dosis von 50 mg/kg (der No-Effect-Level für Entwicklungsparameter lag bei einer Dosis von 25 mg/kg oder 380 mg/m2 mit einer Exposition (AUC), die annähernd 4fach über der beim Menschen zu erwartenden liegt). Ratten, die oral mit Hydromorphon 10 mg/kg (308 mg/m mit einer AUC, die etwa 1,8 mal über der für den Menschen erwarteten liegt) behandelt wurden, zeigten keine foetale Schädigung.
Peri- und postpartal stieg die Mortalität von Rattenbabies (F1) bei 2 und 5 mg/kg/Tag an und das Körpergewicht blieb während der Stillperiode reduziert.
Es wurden keine klinischen Befunde oder Befunde nach Autopsie erhoben, die im Zusammenhang mit der Gabe von Hydromorphon an das Muttertier standen.
Hydromorphon war nicht mutagen im AMES-Test und im Maus-Mikronukleus-Assay.
Außerdem war Hydromorphon im Maus-Lymphoma-Test ohne exogene Metabolisie-rung (S9) ebenfalls nicht mutagen. Unter den Bedingungen exogener Metabolisie-rung war Hydromorphon in Konzentrationen < 100 gg/ml nicht mutagen. Mutagene Eigenschaften konnten in Konzentrationen von > 200 gg/ml beobachtet werden, welche signifikant höher liegen als die erwarteten durchschnittlichen Plasmaspitzenkonzentrationen im Menschen.
Langzeitstudien zur Kanzerogenität wurden nicht durchgeführt.
6. Pharmazeutische Angaben
6.1. Liste der sonstigen Bestandteile
Retardkapsel-Inhalt:
Mikrokristalline Cellulose, Methylhydroxypropylcellulose, Ethylcellulose, gereinigtes Wasser, hochdisperses Siliciumdioxid, Dibutyldecandioat
Retardkapsel-Hülle:
Gelatine, Natriumdodecylsulfat, Titandioxid (E171) zusätzlich bei
4 mg: Erythrosin (E 127), Indigocarmin (E 132),
8 mg: Erythrosin (E 127),
16 mg: Eisenoxidhydrat (E 172)
24 mg: Indigocarmin (E 132)
Markierungstinte:
Schellack, Propylenglycol, Eisenoxid schwarz (E 172).
6.2. Inkompatibilitäten Nicht zutreffend.
6.3. Dauer der Haltbarkeit
2 Jahre
6.4. Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Nicht über 25° C lagern.
In der Originalverpackung aufbewahren.
Den Inhalt vor Feuchtigkeit schützen.
6.5. Art und Inhalt des Behältnisses
Verpackungsart: PVC/PVdC-Durchdrückpackungen mit Aluminiumfolie.
Packungsgrößen: 20, 50 oder 100 Retardkapseln
Klinikpackung: 10 x 10 Retardkapseln
6.6. Besondere Vorsichtsmaßnahmen für die Beseitigung Keine speziellen Hinweise.
7. Inhaber der Zulassung
Mundipharma GmbH Mundipharmastraße 2 65549 Limburg Telefon: (0 64 31) 701-0 Telefax: (0 64 31) 7 42 72
8. Zulassungsnummern
4 mg: 33162.00.00 8 mg: 33162.01.00 16 mg: 33162.02.00
24 mg: 33162.03.00
9. Datum der Erteilung der Zulassung
4 mg: 19.12.1997 8 mg: 19.12.1997 16 mg: 19.12.1997 24 mg: 19.12.1997
10. Stand der Information Februar 2015
11. Verkaufsabgrenzung Verschreibungspflichtig Betäubungsmittel
Weitere Angaben:
Mundipharma Service für Fragen zum Präparat und zur Therapie:
- Gebührenfreie Info-Line: (0800) 8 55 11 11
- E-Mail: medinfo@mundipharma.de
- Internet: http://www.mundipharma.de
Seite 11 /11