Propigen 5 Mg
Seite 1 von 11
ZUSAMMENFASSUNG DER MERKMALE DES ARZNEIMITTELS FACHINFORMATION
1. BEZEICHNUNG DES ARZNEIMITTELS
Propigen 5 mg, überzogene Tabletten
2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
W irkstoff: Propiverinhydrochlorid
1 überzogene Tablette enthält 5 mg Propiverinhydrochlorid, entsprechend 4,55 mg Propiverin.
Sonstige Bestandteile: Eine überzogene Tablette enthält Lactose-Monohydrat (34 mg), GlucoseMonohydrat (0,31 mg), Sucrose (23,7 mg) und Gelborange S (0,2 mg).
Die vollständige Auflistung der sonstigen Bestandteile siehe Abschnitt 6.1.
3. DARREICHUNGSFORM
Überzogene Tablette, gelborange gefärbt, linsenförmig.
4. KLINISCHE ANGABEN
4.1 Anwendungsgebiete
Zur symptomatischen Behandlung von Harninkontinenz und/oder erhöhter Miktionsfrequenz und Harndrang bei Patienten mit überaktiver Blase oder einer neurogenen Detrusorhyperaktivität (Detrusorhyperreflexie) durch Rückenmarkschädigungen.
4.2 Dosierung, Art und Dauer der Anwendung
Überzogene Tabletten zum Einnehmen Empfohlene Tagesdosen:
Kinder: täglich durchschnittlich 0,8 mg/kg Körpergewicht in zwei bis drei Einzeldosen Mögliche Dosierschemen
|
Körpergewicht (kg) |
Propigen 5 mg pro Tag |
|
12 - 16 |
1 - 0 - 1 |
|
17 - 22 |
1 - 1 - 1 |
|
23 - 28 |
2 - 0 - 2 |
|
29 - 34 |
2 - 1 - 2 |
|
> 35 |
2 - 2 - 2 |
|
oder 3 - 0 - 3 |
Bei Kindern mit einem Körpergewicht über 35 kg entspricht die Maximaldosierung der Standarddosierung bei Erwachsenen von zweimal täglich 15 mg (2 x 3 Propigen 5 mg).
Die Behandlung der überaktiven Blase sollte nicht vor dem 5. Lebensjahr beginnen, da in vielen Fällen die organische Entwicklung noch nicht abgeschlossen ist. Die Behandlung der neurogenen Detrusorhyperaktivität aufgrund von Rückenmarkschädigung kann dagegen auch vor dem 5. Lebensjahr beginnen. Die Gabe von Propiverinhydrochlorid an Kinder unter 1 Jahr wird aufgrund fehlender Daten nicht empfohlen.
Die Behandlung von Kindern soll nur im Rahmen eines therapeutischen Gesamtkonzeptes erfolgen (z.B. sog. “Urotherapie” bei idiopathischer Blasenüberaktivität).
Wegen des geringen Wirkstoffgehaltes wird Propigen 5 mg hauptsächlich im Kindesalter bzw. bei Erwachsenen mit geringem Körpergewicht angewendet.
Erwachsene: Als Standarddosis bei idiopathischer Detrusorhyperaktivität werden zweimal täglich 15 mg Propiverinhydrochlorid empfohlen; eine Steigerung auf dreimal täglich ist möglich. Einige Patienten können bereits auf eine Dosis von 15 mg täglich ansprechen (3 x 5 mg).
Bei neurogener Detrusorhyperaktivität wird eine Dosierung von dreimal täglich 15 mg Propiverinhydrochlorid empfohlen. Die maximal empfohlene Tagesdosis ist 45 mg.
Ältere Patienten: Im Allgemeinen gibt es kein spezielles Dosierschema für Ältere (siehe Abschnitt 5.2).
Anwendung bei Patienten mit eingeschränkter Nierenfunktion: Bei Patienten mit einer leichten oder mittelschweren Einschränkung der Nierenfunktion muss die Dosierung nicht angepasst werden, diese sollten jedoch mit Vorsicht behandelt werden. Bei Patienten mit starker Einschränkung der Nierenfunktion (Kreatinin-Clearance < 30 ml/min) beträgt die maximale Tagesdosis 30 mg.
Anwendung bei Patienten mit eingeschränkter Leberfunktion: Bei Patienten mit einer leichten Einschränkung der Leberfunktion besteht keine Notwendigkeit der Dosisanpassung, die Behandlung sollte jedoch mit Vorsicht erfolgen. Studien zur Anwendung von Propiverinhydrochlorid bei Patienten mit mittelschwerer oder schwerer Einschränkung der Leberfunktion wurden nicht durchgeführt. Die Anwendung wird deshalb bei diesen Patienten nicht empfohlen.
Die gleichzeitige Einnahme von Propiverin mit einer fettreichen Mahlzeit erhöht die Bioverfügbarkeit von Propiverin. Die Einnahme sollte deshalb vor den Mahlzeiten erfolgen. Dies ist besonders für Patienten mit Einschränkung von Leber- oder Nierenfunktion von Bedeutung (siehe Abschnitt 5.2).
4.3 Gegenanzeigen
Propigen 5 mg darf bei Patienten mit bekannter Überempfindlichkeit gegen den Wirkstoff oder einen der sonstigen im Abschnitt 6.1 aufgeführten Bestandteile sowie bei Patienten mit einer der folgenden Erkrankungen nicht angewendet werden:
- Darmobstruktion
- ausgeprägte Blasenentleerungsstörungen mit vorhersehbarem Harnverhalt
- Myasthenia gravis
- Darmatonie
- schwere Colitis ulcerosa
- toxisches Megacolon
- unbehandeltes Engwinkelglaukom
- moderate oder ausgeprägte Leberfunktionsstörung
- Tachyarrhythmien.
4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Das Arzneimittel sollte mit Vorsicht angewendet werden bei Patienten mit:
- autonomer Neuropathie
- Nierenfunktionsstörungen (siehe Abschnitt 4.2)
- Leberfunktionsstörungen (siehe Abschnitt 4.2).
Die Symptome folgender Erkrankungen können sich nach Verabreichung des Arzneimittels verstärken:
- schwere kongestive Herzinsuffizienz (NYHA IV)
- Prostatahyperplasie
- Hiatushernie mit Refluxoesophagitis
- Arrhythmie
- Tachykardie.
Wie auch andere Anticholinergika induziert Propiverin eine Mydriasis. Daher kann bei prädisponierten Personen mit engem Kammerwinkel der vorderen Augenkammer ein erhöhtes Risiko bestehen, Glaukomanfälle zu induzieren. Es liegen Berichte vor, dass Wirkstoffe dieser Arzneimittelgruppe Glaukomanfälle induzieren oder verstärken können.
Pollakisurie und Nykturie infolge von Nierenerkrankungen oder dekompensierter Herzinsuffizienz sowie organische Blasenerkrankungen (z. B. Harnwegsinfektionen, Malignome) sollten vor der Behandlung ausgeschlossen werden.
Patienten, die mit potenten Inhibitoren der flavinhaltigen Monooxygenase (FMO), z.B. Methimazol und gleichzeitig mit potenten Inhibitoren des Cytochrom-Enzyms CYP 3A4/5 (z. B. Ketoconazol) behandelt werden, sollten mit der geringst möglichen Anfangsdosis behandelt werden. Die Dosis kann anschließend vorsichtig auftitriert werden (siehe Abschnitte 4.5, 5.2).
Patienten mit der seltenen hereditären Galactose-Intoleranz, Lactase-Mangel, Fructose-Intoleranz, Glucose-Galactose-Malabsorption oder Saccharase-Isomaltase-Mangel sollten Propigen 5 mg nicht einnehmen.
Gelborange S kann allergische Reaktionen auslösen.
Propigen 5 mg ist glutenfrei.
4.5 Wechselwirkungen mit anderen Arzneimittel und sonstige Wechselwirkungen
Wirkungsverstärkung durch gleichzeitige Verabreichung von tricyclischen Antidepressiva (z.B. Imipramin), Tranquilizern (z.B. Benzodiazepine), Anticholinergika, Amantadin, Neuroleptika (z.B. Phenothiazine) und Beta-Adrenozeptor-Agonisten (Beta-Sympathikomimetika). Wirkungsabschwächung durch gleichzeitige Behandlung mit Cholinergika. Blutdrucksenkung bei Patienten unter Isoniazidbehandlung. Die Wirkung von Prokinetika wie Metoclopramid kann verringert werden.
Pharmakokinetische Wechselwirkungen mit anderen Wirkstoffen, die durch Cytochrom P450 3A4 (CYP 3A4) verstoffwechselt werden, sind möglich. Ein sehr ausgeprägter Konzentrationsanstieg wird für solche Wirkstoffe jedoch nicht erwartet, da die Wirkungen von Propiverin im Vergleich zu klassischen Enzyminhibitoren (z. B. Ketoconazol oder Grapefruit-Saft) gering sind. Propiverin gilt als schwacher Hemmer der mikrosomalen Monooxygenase (CYP 3A4). Pharmakokinetische Untersuchungen bei Patienten, die gleichzeitig starke CYP 3A4-Hemmer wie Azol-Antimykotika (z.B. Ketoconazol, Itraconazol) oder Makrolid-Antibiotika (z.B. Erythromycin, Clarithromycin) erhalten, sind nicht durchgeführt worden.
Bei Patienten, die Arzneimittel einnehmen, die wirksame FMO-Hemmer sind, wie z.B. Methimazol, und gleichzeitig mit starken CYP 3A4-Hemmern behandelt werden, sollte die Behandlung mit der geringsten empfohlenen Dosis beginnen. Die Dosierung kann anschließend erhöht werden. Vorsicht ist jedoch geboten, der Arzt sollte diese Patienten sorgfältig bezüglich auftretender Nebenwirkungen überwachen (siehe Abschnitte 4.2, 4.4).
4.6 Fertilität, Schwangerschaft und Stillzeit
Es liegen keine klinischen Daten über die Anwendung von Propiverin bei Schwangeren und Stillenden vor. Tierexperimentelle Studien haben Reproduktionstoxizität gezeigt (siehe Abschnitt 5.3). Propiverin wurde in die Milch laktierender Säugetiere ausgeschieden. Das potentielle Risiko für den Menschen ist nicht bekannt.
Daher darf Propigen 5 mg nicht an schwangere oder stillende Frauen verabreicht werden.
4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Propiverinhydrochlorid kann zu Benommenheit und Verschwommensehen führen. Dadurch kann unter Einnahme dieses Arzneimittels die Fähigkeit des Patienten zum Ausführen von Tätigkeiten, die mentale Aufmerksamkeit erfordern, wie etwa das Führen von Kraftfahrzeugen, das Bedienen von Maschinen oder das Ausführen gefährlicher Arbeiten, eingeschränkt sein.
Beruhigungsmittel können die von Propiverinhydrochlorid verursachte Benommenheit verstärken.
4.8 Nebenwirkungen
Bei der Bewertung von Nebenwirkungen werden folgende Häufigkeiten zugrunde gelegt:
Sehr häufig (> 1/10)
Häufig (> 1/100 bis < 1/10)
Gelegentlich (> 1/1.000 bis < 1/100)
Selten (> 1/10.000 bis < 1/1.000)
Sehr selten (< 1/10.000)
Nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
Psychiatrische Erkrankungen
Sehr selten: Verwirrtheit, Unruhe
Nicht bekannt: Halluzinationen
Erkrankungen des Nervensystems
Häufig: Kopfschmerzen
Gelegentlich: Tremor, Schwindel, Geschmacksstörungen
Augenerkrankungen
Häufig: Akkommodation anormal, Akkommodationsstörungen, anormaler Visus
Herzerkrankungen
Sehr selten: Palpitationen
Gefäßerkrankungen
Gelegentlich: Blutdrucksenkung mit Benommenheit, Erröten
Erkrankungen des Gastrointestinaltraktes
Sehr häufig: Mundtrockenheit
Häufig: Obstipation, Bauchschmerzen und Dyspepsie
Gelegentlich: Übelkeit/Erbrechen
Erkrankungen der Haut und des Unterhautzellgewebes
Selten: Ausschlag bei Idiosynkrasie (Propiverinhydrochlorid) oder Überempfindlichkeit
(Hilfsstoffe, z. B. Farbstoff)
Erkrankungen der Nieren und Harnwege
Gelegentlich: Hamverhalt
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Häufig: Müdigkeit und Erschöpfung
Kinder und Jugendliche
In Studien mit Kindern sind außerdem folgende Nebenwirkungen berichtet wurden: Appetitlosigkeit, Schlafstörungen und Konzentrationsstörungen.
Alle unerwünschten Wirkungen sind vorübergehend und klingen nach einer Dosisreduzierung oder bei Beendigung der Therapie nach maximal 1 - 4 Tagen ab.
Bei einer Langzeittherapie sollten die Leberenzyme kontrolliert werden, da in seltenen Fällen reversible Leberenzymveränderungen auftreten können. Bei Patienten mit der Gefahr einer Glaukomentwicklung wird die Kontrolle des Augeninnendrucks empfohlen.
Bei Harnwegsinfekten sollte besonders auf die Restharnmenge geachtet werden.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Arzneimittel und Medizinprodukte, Abt. Pharmakovigilanz, Kurt-Georg-Kiesinger Allee 3, D-53175 Bonn, Website: www.bfarm.de anzuzeigen.
4.9 Überdosierung
Eine Überdosierung mit dem Muskarinrezeptor-Antagonisten Propiverinhydrochlorid kann potentiell zu zentralnervösen anticholinergen Effekten, wie z. B. Unruhe, Schwindel, Übelkeit, Sprach- und Sehstörungen und Muskelschwäche, führen. Darüber hinaus können schwere Trockenheit der Schleimhäute, Tachykardie und Harnverhalt auftreten.
Die Behandlung der Überdosierung kann das Auslösen von Erbrechen oder eine Magenspülung unter Verwendung eines eingeölten Schlauches beinhalten (Vorsicht: trockene Schleimhäute!). Dann ist symptomatisch wie bei Atropin-Überdosierung, z. B. Physostigmin mit einer Dosierung von 1,0 bis
2,0 mg bei Erwachsenen durch langsame intravenöse Injektion (kann bei Bedarf bis zu einer Gesamtmenge von 5 mg wiederholt werden), zu behandeln.
Ein 14-jähriges Mädchen, das 450 mg Propiverinhydrochlorid eingenommen hatte, zeigte eine Konfabulation. Die Jugendliche erholte sich vollständig.
5. PHARMAKOLOGISCHE EIGENSCHAFTEN
5.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe Urologische Spasmolytika
ATC-Code
G04B D06
Wirkungsweise
Muskulotrope Spasmolyse durch Hemmung des Calcium-Einstroms und Modulation des intrazellulären Calciums in der glatten Muskulatur der Harnblase.
Hemmung der efferenten Bahnen des Nervus pelvicus durch anticholinerge Wirkung.
Pharmakodynamische Wirkungen
Am Tier bewirkt Propiverinhydrochlorid eine dosisabhängige Abnahme des intravesikalen Drucks und eine Erhöhung der Blasenkapazität. Der Effekt beruht auf der Summe der pharmakologischen Eigenschaften von Propiverin und drei aktiven Harnmetaboliten, wie an isolierten Detrusorstreifen von Mensch und Tier gezeigt werden konnte.
In einer randomisierten, placebo-kontrollierten, doppelblinden Phase-III-Studie konnte eine signifikante Wirksamkeit (Abnahme der Inkontinenzepisoden und der Miktionsfrequenz, Zunahme des Miktionsvolumens) von Propiverin an Kindern belegt werden.
5.2 Pharmakokinetische Eigenschaften
Die folgenden Informationen beziehen sich auf eine Formulierung mit 15 mg Propiverinhydrochlorid. Allgemeine Eigenschaften der aktiven Substanz
Propiverin wird fast vollständig aus dem Magen-Darm-Trakt resorbiert. Es unterliegt einem extensiven First-Pass-Metabolismus. Wirkungen an den Zellen der glatten Muskulatur der Harnblase sind durch den Wirkstoff sowie durch drei aktive Metaboliten bedingt, die schnell in den Urin ausgeschieden werden.
Resorption
Nach oraler Gabe von Propiverinhydrochlorid 15 mg wird Propiverin schnell aus dem Gastrointestinaltrakt resorbiert und erreicht maximale Plasmakonzentrationen nach 2,3 Stunden.
Die mittlere absolute Bioverfügbarkeit von Propiverinhydrochlorid 15 mg beträgt 40,5 % (arithmetischer Mittelwert aus AUC0-<x> (p.o.) / AUC0-<x> (i.v.)).
Durch Nahrungsaufnahme wird die Bioverfügbarkeit von Propiverin erhöht (mittlerer Anstieg um das 1,3-fache), jedoch ohne signifikanten Einfluss auf die maximale Plasmakonzentration von Propiverin oder seines Hauptmetaboliten Propiverin-N-Oxid. Es ist unwahrscheinlich, dass dieser Unterschied in der Bioverfügbarkeit klinische Bedeutung hat. Eine Dosisanpassung könnte jedoch für Patienten mit eingeschränkter Leber- und/oder Nierenfunktion notwendig sein. Deshalb sollte Propiverin von allen Patienten vor dem Essen eingenommen werden.
Verteilung
Nach Gabe von Propiverinhydrochlorid 15 mg t. i. d. wird ein Steady state (Fließgleichgewicht) nach 4 bis 5 Tagen auf einem höheren Konzentrationsniveau als nach einer Einzelgabe erreicht (CMittelwert = 61 ng/ml).
Das Verteilungsvolumen wurde bei 21 gesunden Probanden nach intravenöser Gabe von Propiverinhydrochlorid bestimmt und lag zwischen 125 und 473 l (Mittelwert 279 l), was darauf hindeutet, dass eine große Menge des verfügbaren Propiverins in periphere Kompartimente verteilt wird. Die Plasmaprotein-Bindung beträgt 90 - 95 % für die Stammverbindung und etwa 60 % für den Hauptmetaboliten.
Plasmakonzentrationen von Propiverin bei 16 gesunden Probanden nach einmaliger und wiederholter Gabe von Propiverin Hydrochlorid 15 mg Überzogene Tabletten (t. i. d. über 6 Tage):
[ng/ml]
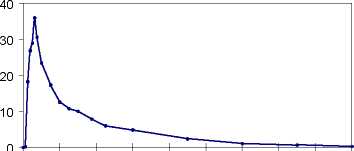
0 8 16 24 32 40 48 56 64 72
Zeit [h]
[ng/ml]
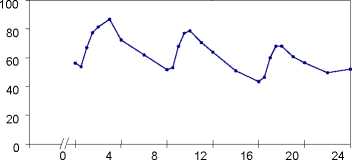
Zeit [h]
Steady-state-Eigenschaften von Propiverin nach wiederholter Gabe von Propiverin Hydrochlorid 15 mg Überzogene Tabletten an 16 gesunde Probanden (t. i. d. über 6 Tage):
|
Dosierungs intervall [hl |
AUC0.x [ng-h/mll |
CV [%l |
PTF [%l |
CV [%l |
CMittelwert [ng/mll |
CV [%l |
|
0 - 8 |
515 |
35 |
57 |
16 |
64 |
36 |
|
8 - 16 |
460 |
33 |
70 |
25 |
57 |
33 |
|
16 - 24 |
421 |
36 |
52 |
39 |
52 |
36 |
|
CV: Variationskoeffizient PTF: Maximum-Minimum-Streuung | ||||||
Biotransformation
Propiverin wird extensiv durch intestinale und hepatische Enzyme metabolisiert. Der Hauptabbauweg schließt die Oxidation des Piperidyl-N ein und wird durch CYP 3A4 und die Flavin-Monooxygenasen (FMO) 1 und 3 vermittelt und führt zur Bildung des weit weniger aktiven N-Oxids, dessen Plasmakonzentration die der Ausgangssubstanz deutlich übersteigt. Vier Metaboliten wurden im Urin nachgewiesen, zwei davon sind pharmakologisch aktiv und können zur therapeutischen Wirksamkeit von Propiverin beitragen.
In vitro ist eine geringfügige Hemmung von CYP 3A4 und CYP 2D6 messbar, die bei Konzentrationen auftritt, die die therapeutischen Plasmakonzentrationen um das 10- bis 100-fache übersteigen (siehe Abschnitt 4.5).
Elimination
Nach oraler Gabe von 30 mg 14C-Propiverinhydrochlorid an gesunde Probanden wurden innerhalb von 12 Tagen 60 % der Radioaktivität im Urin und 21 % der Radioaktivität in den Faeces gefunden. Weniger als 1 % einer oralen Gabe wird unverändert mit dem Urin ausgeschieden. Die mittlere totale Clearance nach einmaliger Gabe von 30 mg beträgt 371 ml/min (191 - 870 ml/min).
Im Rahmen von drei Studien, in die insgesamt 37 gesunde Probanden eingeschlossen waren, wurden mittlere Eliminationshalbwertzeiten von 14,1 bzw. 20,1 bzw. 22,1 Stunden ermittelt.
Linearität/Nichtlinearität
Die pharmakokinetischen Parameter von Propiverin und Propiverin-N-Oxid nach oraler Gabe von 10 - 30 mg Propiverinhydrochlorid stehen in einem linearen Zusammenhang mit der Dosis.
Während des Steady state sind keine Veränderungen in der Pharmakokinetik im Vergleich zur Einzelgabe zu sehen.
Eigenschaften bei Patienten
Nierenfunktionseinschränkung
Aus einer Single-Dose-Studie an 12 Patienten mit einer Kreatininclearance < 30 ml/min wurde.geschlussfolgert, dass eine schwere Nierenfunktionseinschränkung die Eliminierung von Propiverin und seinem Hauptmetaboliten Propiverin-N-Oxid nicht wesentlich ändert. Eine Dosisanpassung ist nicht notwendig, wenn die Gesamt-Tagesdosis 30 mg Propiverinhydrochlorid nicht überschreitet. Soll eine höhere Dosis gegeben werden, ist eine vorsichtige Titration der Dosis unter Berücksichtigung der anticholinergen Wirkungen als Marker für die Verträglichkeit zu empfehlen.
Leberinsuffizienz
Verglichen mit 12 gesunden Kontrollpersonen war die Steady-state-Pharmakokinetik von Propiverinhydrochlorid 15 mg t. i. d. über 5 Tage ähnlich der von 12 Patienten mit milder bis moderater Leberfunktionseinschränkung durch eine Fettleber. Für schwere Leberfunktionseinschränkungen liegen keine Daten vor.
Alter
Der Vergleich von Tal-Plasmakonzentrationen während des Steady state (Mictonorm® t. i. d. über 28 Tage) zeigt keinen Unterschied zwischen älteren Patienten (60 - 85 Jahre; Mittelwert 68) und jungen gesunden Probanden. Das Verhältnis der Muttersubstanz zum Metaboliten bleibt bei älteren Patienten unverändert, was darauf hindeutet, dass die metabolische Umsetzung von Propiverin in seinen Hauptmetaboliten Propiverin-N-Oxid nicht altersabhängig oder nicht limitierend bei der Gesamtausscheidung ist.
Kinder und Jugendliche
Eine Dosis-Findungs-Studie an Kindern belegte für die Dosierung von zweimal täglich durchschnittlich 0,4 mg/kg Körpergewicht ein ausgewogenes Verhältnis zwischen Wirksamkeit und Verträglichkeit. Bis zum empfohlenen Dosierungsbereich sind die pharmakokinetischen Eigenschaften (z. B. AUC0-8, cmax, cav) dosisproportional. Nach Gabe einer Dosis von zweimal täglich 0,4 mg/kg Körpergewicht erreichen die Serumspiegel bei Kindern im Alter von 5 bis 10 Jahren in etwa die Werte wie nach Gabe der therapeutischen Dosis von zweimal täglich 15 mg Propiverinhydrochlorid bei Erwachsenen.
Glaukom-Patienten
Wie in zwei Placebo-kontrollierten Studien gezeigt werden konnte, erhöht Propiverinhydrochlorid 15 mg t. i. d. über 7 Tage nicht den Augeninnendruck bei Patienten mit Weitwinkelglaukom und bei Patienten mit behandeltem (kontrolliertem) Engwinkelglaukom.
5.3 Präklinische Daten zur Sicherheit
In Langzeituntersuchungen mit oraler Gabe an zwei Säugetierarten waren die wichtigsten behandlungsbezogenen Effekte Veränderungen in der Leber (einschließlich Erhöhung der Leberenzyme). Diese waren durch Leberhypertrophie und -verfettung gekennzeichnet. Die Verfettung war nach Abbruch der Behandlung reversibel.
In Untersuchungen an Tieren kam es bei hochdosierter oraler Verabreichung des Wirkstoffes an trächtige Weibchen zu einer verzögerten Skelettentwicklung bei den Nachkommen. Bei laktierenden Säugetieren wurde Propiverinhydrochlorid in die Milch ausgeschieden.
Es wurde kein Hinweis auf Mutagenität gefunden. Eine Karzinogenitätsstudie an Mäusen zeigte im hohen Dosisbereich eine erhöhte Inzidenz an hepatozellulären Adenomen und Karzinomen bei männlichen Tieren. In einer Karzinogenitätsstudie an Ratten traten im hohen Dosisbereich bei männlichen Tieren hepatozelluläre Adenome, Nierenadenome und Harnblasenpapillome auf, während bei weiblichen Tieren im hohen Dosisbereich Endometriumpolypen auftraten. Die Tumoren wurden jedoch alle als artspezifisch und daher nicht als klinisch relevant eingeschätzt.
6. PHARMAZEUTISCHE EIGENSCHAFTEN
6.1 Liste der sonstigen Bestandteile
Tablettenkern: Lactose-Monohydrat, Cellulosepulver, Magnesiumstearat (Ph.Eur., pflanzlich)
Tablettenhülle: Calciumcarbonat, Gelborange S (E 110), Glucose-Monohydrat (Ph.Eur.), Arabisches Gummi, Macrogol 6000, Montanglycolwachs, Sucrose (Saccharose (Ph.Eur.)), Hochdisperses Siliciumdioxid, Talkum, Titandioxid E 171, Weißer Ton.
6.2 Inkompatibilitäten
Nicht zutreffend.
6.3 Dauer der Haltbarkeit
5 Jahre
6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Dieses Arzneimittel erfordert keine besonderen Lagerungsbedingungen.
6.5 Art und Inhalt des Behältnisses
PVC/Aluminium-Blister in Faltschachteln mit 28, 30, 49, 50, 98 und 100 überzogenen Tabletten.
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung <
7. KEINE BESONDEREN ANFORDERUNGENINHABER DER ZULASSUNG
APOGEPHA Arzneimittel GmbH
Kyffhäuserstr. 27
01309 Dresden
Tel.: 03 51 / 3 36 33
Fax: 03 51/ 3 36 34 40
8. ZULASSUNGSNUMMER(N)
74126.00.00
9. DATUM DER ERTEILUNG DER ZULASSUNG/VERLÄNGERUNG DER ZULASSUNG
22.10.2012
10. STAND DER INFORMATION
Oktober 2014
11. VERKAUFSABGRENZUNG
V erschreibungspflichtig