Recombinate Antihaemophilie Faktor (Rekombinant) 500
alt informationenFachinformation
(Zusammenfassung der Merkmale des Arzneimittels)
1. BEZEICHNUNG DES ARZNEIMITTELS
Recombinate Antihämophilie Faktor (rekombinant) 500 Pulver und Lösungsmittel zur Herstellung einer Injektionslösung
2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
Octocog alfa 50 I.E. pro ml der rekonstituierten Lösung
Nach Rekonstitution: Eine Durchstechflasche mit 10 ml enthält 500 I.E. Octocog
alfa
Recombinate Antihämophilie Faktor (rekombinant) 500 enthält nominal 500 I.E. Octocog alfa, rekombinanten Gerinnungsfaktor VIII, pro Durchstechflasche.
Das gebrauchsfertige Produkt enthält pro ml etwa 50 I.E. Octocog alfa, rekombinanten Gerinnungsfaktor VIII, wenn es mit 10 ml sterilisiertem Wasser für Injektionszwecke rekonstituiert wurde.
Die Stärke (I.E.) wird mit dem chromogenen Test gemäß der Europäischen Pharmakopoe gegen den FDA Mega-Standard bestimmt, der gegen den WHO-Standard kalibriert ist. Die spezifische Aktivität von Recombinate Antihämophilie Faktor (rekombinant) 500 beträgt ungefähr 4000 - 8000 I.E./mg Protein.
Recombinate Antihämophilie Faktor (rekombinant) 500 enthält rekombinanten Gerinnungsfaktor VIII (INN: Octocog alfa). Octocog alfa (rekombinanter Gerinnungsfaktor VIII) ist ein gereinigtes Protein, das aus 2.332 Aminosäuren besteht. Seine Aminosäurensequenz ist vergleichbar mit Faktor VIII, und die post-translationalen Modifikationen ähneln dem plasmatischen Molekül. Rekombinanter Gerinnungsfaktor VIII ist ein Glykoprotein, das durch eine genetisch veränderte Ovarialzell-Linie des Chinesischen Hamsters produziert wird.
Sonstige Bestandteile: Die vollständige Auflistung der sonstigen Bestandteile siehe Abschnitt 6.1.
3. DARREICHUNGSFORM
Pulver und Lösungsmittel zur Herstellung einer Injektionslösung.
Weißes bis cremefarbenes, bröckeliges Pulver. Das Lösungsmittel (sterilisiertes Wasser für Injektionszwecke) ist eine klare und farblose Lösung.
4. KLINISCHE ANGABEN
4.1 Anwendungsgebiete
Behandlung und Prophylaxe von Blutungen bei Patienten mit Hämophilie A (angeborener Faktor VIII-Mangel).
Das Produkt enthält keinen von-Willebrand-Faktor und eignet sich daher nicht zur Behandlung des von-Willebrand-Jürgens-Syndroms.
Recombinate Antihämophilie Faktor (rekombinant) 500 eignet sich für alle Altersklassen vom Neugeborenen bis zu Erwachsenen.
4.2 Dosierung, Art und Dauer der Anwendung
Dosierung und Dauer der Substitutionstherapie hängen von der Schwere der Hämostasestörung sowie dem Ort und Ausmaß der Blutung und dem klinischen Zustand des Patienten ab. Die Behandlung sollte in Zusammenarbeit mit einem in Hämostasestörungen erfahrenen Arzt und mit einem Labor erfolgen, das in der Lage ist, AHF im Plasma zu bestimmen.
Die verabreichten Faktor VIII-Einheiten werden in Internationalen Einheiten (I.E.) angegeben, die sich auf den aktuellen Standard der WHO für Faktor VIII-Produkte beziehen. Die Faktor VIII-Aktivität im Plasma wird entweder in Prozent (bezogen auf Normalplasma) ausgedrückt oder in Internationalen Einheiten (bezogen auf einen Internationalen Faktor VIII-Standard im Plasma). Eine Internationale Einheit (I.E.) an Faktor VIII-Aktivität entspricht der Menge an Faktor VIII in einem ml normalen, menschlichen Plasmas.
Der nach der Gabe von Recombinate Antihämophilie Faktor (rekombinant) 500 zu erwartende in v/Vo-Anstieg des Faktor VIII-Plasmaspiegels in I.E./dl Plasma oder % (Prozent) des Normalwertes kann annähernd geschätzt werden, indem man die verabreichte Dosis pro kg Körpergewicht (I.E./kg) mit zwei multipliziert.
Die Berechnungsmethode wird anhand der folgenden Beispiele aufgezeigt.
Erwarteter Faktor VIII-Anstieg in % = Anzahl d. verabr. Einheiten x 2 % / I.E./ kg
Körpergewicht (kg)
Beispiel für einen 70 kg schweren Erwachsenen: 1750 I.E. x 2 % / I.E./ kg = ~50 %
70 kg
oder
erforderliche Dosis (I.E.): Körpergewicht (kg) x erwünschter Faktor VIII-Anstieg in %
2 % / I.E. / kg
Beispiel für ein 40 kg schweres Kind: 40 kg x 70 % = 1400 I.E.
2% / I.E. / kg
Die sorgfältige Kontrolle der Substitutionstherapie ist besonders bei größeren, chirurgischen Eingriffen oder lebensbedrohlichen Blutungen von größter Wichtigkeit. Obwohl die Dosis mit Hilfe der obigen Formeln ungefähr abgeschätzt werden kann, wird, wann immer möglich, dringend empfohlen, in geeigneten Zeitabständen entsprechende Laboruntersuchungen einschließlich regelmäßiger Bestimmung der AHF-Aktivität im Patientenplasma durchzuführen, um sicherzustellen, dass ausreichende AHF-Spiegel erreicht und aufrechterhalten werden. Wird die erwartete AHF-Konzentration im Patientenplasma nicht erreicht, oder kommt die Blutung auch nach einer angemessenen Dosis nicht zum Stillstand, so ist vom Vorhandensein eines Inhibitors auszugehen. Mit Hilfe entsprechender Laborverfahren kann ein Inhibitor nachgewiesen und quantifiziert werden. Die Maßeinheit ist definiert als AHF in Internationalen Einheiten, der pro ml Plasma (Be-thesda-Einheiten) oder durch das gesamte, geschätzte Plasmavolumen neutralisiert wird. Bei einem Inhibitortiter von weniger als 10 Bethesda-Einheiten pro ml kann der Inhibitor durch eine entsprechend höhere Dosierung von AHF neutralisiert werden. Danach sollte die Verabreichung weiterer Internationaler Einheiten von AHF zur erwarteten Wirkung führen. In dieser Situation ist die gerinnungsanalytische Kontrolle des AHF-Spiegels unerlässlich. Inhibitortiter von über 10 Bethesda-Einheiten pro ml können eine Kontrolle der Hämostase durch Faktor VIII-Gaben unmöglich machen, da eine sehr große Dosis erforderlich wäre.
Das folgende Dosierungsschema in Tabelle 1 kann als Richtlinie für Erwachsene und Kinder verwendet werden. Dosis und Anwendungshäufigkeit sollten sich immer nach der klinischen Wirksamkeit im Einzelfall richten.
Recombinate Antihämophilie Faktor (rekombinant) 500 kann auch zur Blutungsprophylaxe (über einen kurzen oder längeren Zeitraum) verwendet werden; die Indikation hierzu ist vom Arzt für den Einzelfall zu stellen.
Tabelle 1: Dosierungsschema bei Blutungen
|
Tabelle 1: Dosierungsschema | ||
|
Blutung | ||
|
Schweregrad der Blutung / Art des chirurgischen Eingriffs |
Erforderlicher Faktor VIII- Plasmaspiegel (% des Normalwertes oder I.E/dl) |
Häufigkeit der Anwendung |
|
Beginnende Gelenkblutungen, Muskelblutungen oder Blutungen im Mund |
20 - 40 |
Infusion alle 12 - 24 Stunden über 1 bis 3 Tage, bis die Blutung (angezeigt durch Schmerzen) steht oder Wundheilung erreicht ist. |
|
Ausgeprägtere Gelenkblutungen, Muskelblutungen oder Hämatome |
30 - 60 |
Infusion alle 12 - 24 Stunden wiederholen; normalerweise 3 Tage lang oder länger, bis Schmerzen und Behinderungen beseitigt sind. |
|
Lebensbedrohliche Blutungen wie intrakraniale Blutungen, Blutungen in den Hals, starke abdominelle Blutungen |
60 - 100 |
Infusion alle 8 - 24 Stunden wiederholen, bis die Gefahr für den Patienten vorüber ist. |
|
Chirurgische Eingriffe | ||
|
Kleinere Eingriffe, einschließlich Zahnextraktion |
30 - 60 |
Einzelinfusion plus orale antifibrinolytische Therapie innerhalb 1 Stunde ist bei ca. 70 % der Fälle ausreichend. Alle 24 Stunden, für mindestens 1 Tag, bis die Wundheilung erreicht ist. |
|
Größere Eingriffe |
80 - 100 (prä- und postoperativ) |
Infusion alle 8 - 24 Stunden -je nach Stand der Wundheilung - wiederholen. |
Bei Patienten mit der erwarteten Halbwertszeit für Faktor VIII entspricht dies dem Spitzenwert der AHF-Aktivität. Falls erforderlich, sollte der Spitzenwert eine halbe Stunde nach der Verabreichung des Medikaments ermittelt werden. Bei Patienten mit relativ kurzer Halbwertszeit für Faktor VIII kann es erforderlich sein, die Dosis und/oder die Verabreichungshäufigkeit zu erhöhen.
Auf jeder Durchstechflasche Recombinate Antihämophilie Faktor (rekombinant) 500 ist die Aktivität des (rekombinanten) Antihämophilie Faktors in I.E. pro Durchstechflasche angegeben.
Diese Stärke bezieht sich auf den Internationalen Standard für Faktor VIII-C-Konzentrat der WHO. Versuche haben gezeigt, dass diese Bestimmungen in
Plastikröhrchen und mit Plastikpipetten durchgeführt werden sollten, und dass Substrat mit einem normalen Wert an von-Willebrand-Faktor verwendet werden sollte, um exakte Aktivitätswerte zu erzielen.
Zur Langzeitprophylaxe gegen Blutungen bei Patienten mit schwerer Hämophilie A werden üblicherweise Dosen zwischen 20 und 40 I.E. Faktor VIII pro kg Körpergewicht in Abständen von 2 bis 3 Tagen gegeben.
Die Patienten sollten hinsichtlich der Entwicklung eines Faktor VIII-Inhibitors überwacht werden. Wenn die erwarteten Faktor VIII-Plasmaspiegel nicht erreicht werden, oder wenn die Blutung mit einer entsprechenden Dosis nicht kontrolliert werden kann, sollte ein Test zur Bestimmung eines Faktor VIII-Inhibitors vorgenommen werden. Bei Patienten mit hohen Inhibitor-Werten kann die Faktor VIII-Therapie unwirksam sein, und es sollten andere therapeutische Maßnahmen in Betracht gezogen werden. Die Behandlung solcher Patienten sollte von Ärzten vorgenommen werden, die Erfahrung in der Hämophilie-Behandlung haben. Siehe auch Abschnitt 4.4.
Kinder und Jugendliche
Recombinate Antihämophilie Faktor (rekombinant) 500 ist geeignet für die Anwendung bei Kindern jeden Alters, einschließlich Neugeborenen (die Sicherheit und Wirksamkeit wurde in Studien sowohl an vorher behandelten als auch an vorher unbehandelten Kindern belegt; siehe Abschnitt 5.1). Bei der Bedarfstherapie unterscheidet sich die Dosierung bei Kindern und Jugendlichen nicht von der von Erwachsenen. Zur Langzeitprophylaxe von Blutungen bei Patienten mit schwerer Hämophilie A können in manchen Fällen kürzere Dosierungsintervalle oder höhere Dosen als üblich erforderlich sein. Die üblichen Dosen liegen zwischen 20 bis 40 I.E. Faktor VIII pro kg Körpergewicht im Abstand von 2 bis 3 Tagen.
Art der Anwendung
Das Präparat wird nach dem Auflösen in dem mitgelieferten Lösungsmittel intravenös verabreicht (siehe auch Abschnitt 6.6). Die rekonstituierte Lösung nicht kühlen. Es wird empfohlen, Recombinate Antihämophilie Faktor (rekombinant) 500 bei Raumtemperatur innerhalb von 3 Stunden nach dem Auflösen zu verabreichen. Die Infusionsgeschwindigkeit, bis hin zu einer maximalen Geschwindigkeit von 10 ml/Min, richtet sich nach dem Wohlbefinden des Patienten. Vor und während der Verabreichung von Recombinate Antihämophilie Faktor (rekombinant) 500 sollte die Pulsfrequenz überprüft werden. Bei einer signifikanten Pulserhöhung klingen die Symptome durch Verlangsamen oder kurzes Unterbrechen der Infusion normalerweise schnell wieder ab (siehe Abschnitte 4.4 und 4.8).
Hinweise zur Rekonstitution des Arzneimittels vor Verabreichung siehe Abschnitt 6.6.
4.3 Gegenanzeigen
Überempfindlichkeit gegen den wirksamen Bestandteil oder einen der sonstigen Bestandteile. Bekannte Allergie gegen Rinder-, Maus- oder Hamsterprotein.
4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Es wurde über schwere allergische Reaktionen bei Recombinate Antihämophilie Faktor (rekombinant) 500 berichtet. Die Patienten sollten über frühe Anzeichen von Überempfindlichkeitsreaktionen wie Nesselausschlag, generalisierte Urtikaria, Engegefühl in der Brust, Stenoseatmung, niedriger Blutdruck und anaphylaktischer Schock informiert werden. Beim Auftreten allergischer oder anaphylaktischer Reaktionen muss die Injektion/Infusion umgehend abgebrochen werden. Geeignete Einrichtungen zur Schockbehandlung sollten vorhanden sein.
Sollten die erwarteten AHF-Konzentrationen im Plasma nicht erreicht werden, oder sollten die Blutungen nach Gabe der entsprechenden Dosis nicht unter Kontrolle gebracht werden, sind geeignete Laboruntersuchungen durchzuführen, um einen eventuell vorhandenen Inhibitor nachzuweisen.
Die Bildung neutralisierender Antikörper (Inhibitoren) gegen Faktor VIII ist eine bekannte Komplikation bei der Behandlung von Hämophilie A-Patienten. Diese Inhibitoren sind normalerweise IgG-Immunglobuline, die gegen die prokoagulato-rische Aktivität des Faktors VIII gerichtet sind und deren inhibitorische Aktivität mit Hilfe des modifizierten Bethesda-Tests in Bethesda-Einheiten (B.E.) pro ml Plasma ausgedrückt wird. Das Risiko einer Bildung von Inhibitoren hängt mit der Expositionsdauer gegenüber dem Faktor VIII zusammen, wobei dieses Risiko innerhalb der ersten 20 Expositionstage am höchsten ist und mit anderen genetischen und umweltbedingten Faktoren zusammenhängt. Selten können sich Inhibitoren nach den ersten 100 Expositionstagen entwickeln. Bei vorbehandelten Patienten mit mehr als 100 Expositionstagen und Hemmkörperbildung in der Vergangenheit wurden beim Wechsel von einem rekombinanten Faktor VIII-Produkt zu einem anderen in einigen Fällen erneut (niedrigtitrige) Hemmkörper beobachtet.
Patienten, die mit rekombinantem Blutgerinnungsfaktor VIII behandelt werden, sollten klinisch und durch Labortests sorgfältig auf die Entwicklung eines Inhibitors hin überwacht werden. Siehe auch Abschnitt 4.8.
Im Interesse des Patienten wird dringend empfohlen, bei jeder Verabreichung von Recombinate Antihämophilie Faktor (rekombinant) 500 die Bezeichnung des Produktes und Chargennummer zu notieren.
Das Arzneimittel enthält 1,5 mmol Natrium pro Durchstechflasche. Dies muss bei Patienten, die unter einer natriumkontrollierten Diät stehen, in Betracht gezogen werden.
Kinder und Jugendliche
Die besonderen Warnhinweise und Vorsichtsmaßnahmen für die Anwendung bei pädiatrischen Patienten unterscheiden sich nicht von denen bei Erwachsenen.
4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Es wurden keine Wechselwirkungsstudien mit Recombinate Antihämophilie Faktor (rekombinant) 500 durchgeführt.
4.6 Fertilität, Schwangerschaft und Stillzeit
Mit Faktor VIII wurden keine Reproduktionsstudien an Tieren durchgeführt. Aufgrund des seltenen Auftretens von Hämophilie A bei Frauen liegen keine Erfahrungen über die Anwendung von Faktor VIII bei Schwangeren und stillenden Müttern vor. Daher sollte Faktor VIIII bei Schwangeren und während der Stillzeit nur dann angewandt werden, wenn dies unbedingt erforderlich ist.
4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Es wurden keine Auswirkungen auf die Fahrtüchtigkeit oder das Bedienen von Maschinen beobachtet.
4.8. Nebenwirkungen
Tabellarische Auflistung von Nebenwirkungen
Die folgende Tabelle führt Nebenwirkungsreaktionen aus Spontanberichten und Klinischen Studien auf. Innerhalb jeder Gruppe werden die unerwünschten Wirkungen nach abnehmendem Schweregrad angegeben.
Die Häufigkeit wurde nach folgenden Kriterien ermittelt: sehr häufig (>1/10), häufig >1/100 bis <1/10), gelegentlich (>1/1.000 bis <1/100), selten (>1/10.000 bis <1/1.000), sehr selten (<1/10.000) und nicht bekannt (d. h. kann aus den verfügbaren Daten nicht eruiert werden).
|
Systemorganklassen gemäß MeDRA-Datenbank |
Häufigkeiten |
Bevorzugter Begriff in der MeDRA-Datenbank |
|
Infektionen und parasitäre Erkrankungen |
gelegentlich |
Ohrinfektionen |
|
Erkrankungen des Blutes und Lymphsystems |
häufig |
Faktor VIII- Hemmung1 |
|
Erkrankungen des Immunsystems |
nicht bekannt |
anaphylaktische Reaktion Überempfindlichkeitsreaktion2 |
|
Erkrankungen des Nervensystems |
gelegentlich |
Schwindelanfälle Tremor |
|
nicht bekannt |
Bewusstseinsverlust Synkopen Kopfschmerz Parästhesien |
|
Herzerkrankungen |
nicht bekannt |
Zyanose Herzrasen |
|
Gefäßerkrankungen |
gelegentlich |
Nasenbluten flüchtige Hautrötungen Blutergüsse Bluthochdruck Blässe peripheres Kältegefühl |
|
Erkrankungen der Atemwege, des Brustraums und des Mediastinums |
gelegentlich |
Schmerzen im Hals und Rachenraum |
|
nicht bekannt |
Atemnot Husten keuchende Atmung | |
|
Erkrankungen des Gastrointestinaltrakts |
gelegentlich |
Übelkeit |
|
nicht bekannt |
Erbrechen Bauchschmerzen | |
|
Erkrankungen der Haut und des Unterhautzellgewebes |
gelegentlich |
übermäßige Schweißproduktion Juckreiz Hautrötung rote Quaddeln |
|
nicht bekannt |
Angioödem Nesselsucht Hautablösungen Hautrötung | |
|
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen |
gelegentlich |
Schmerzen in den Extremitäten |
|
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort |
häufig |
Schüttelfrost |
|
gelegentlich |
Müdigkeit Fieber | |
|
nicht bekannt |
Unwohlsein Reaktionen an der Injektionsstelle Brustschmerzen Engegefühl in der Brust | |
|
Untersuchungen |
gelegentlich |
abnormale Hörtests |
1
Bei der klinischen PTP-Studie (PTP = vorbehandelte Patienten) entwickelte keiner der 71 Patienten von sich aus Faktor VIII-Antikörper, aber bei 22 von 723 durch ein Protokoll auswertbaren PUPs (PUP = unbehandelte Patienten) zeigten sich Faktor VIII-Antikörper. Patienten, die mit Recombinate Antihämophilie Faktor (rekombinant) 500 vorbehandelt worden waren, entwickelten Faktor VIII-Antikörper und ihre Häufigkeitsangabe basiert auf den o. g. PUP-Daten. 10 der genannten 22 Patienten waren hochtitrig (>5 Bethesda-Einheiten), und 12 waren niedrigtitrig (<5 Bethesda-Einheiten).
2
Frühe Anzeichen von Überempfindlichkeitsreaktionen, sind z. B. Nesselsucht, Atemnot, Husten, Brustbeschwerden, keuchende Atmung, Anaphylaxie, Rötung, Blutdruckabfall, Juckreiz, Schüttelfrost, flüchtige Hautrötungen, Fieber, Zyanose, Herzrasen, Übelkeit, Synkopen und Kopfschmerz. Vorsicht wird angeraten bei Patienten mit bekannten, allergischen Reaktionen auf Bestandteile des Präparates (siehe Abschnitte 4.3 und 4.4)
Beschreibung einzelner Nebenwirkungen
Die Bildung neutralisierender Antikörper (Inhibitoren) gegen Faktor VIII ist eine bekannte Komplikation bei der Behandlung von Hämophilie A-Patienten. Diese Inhibitoren sind unweigerlich IgG-Immunglobuline, die gegen die prokoagulatori-sche Aktivität des Faktors VIII gerichtet sind und deren inhibitorische Aktivität mit Hilfe des modifizierten Bethesda-Tests in Bethesda-Einheiten (B.E.) pro ml Plasma ausgedrückt wird.
Das Risiko einer Bildung von Inhibitoren hängt mit der Expositionsdauer gegenüber dem Faktor VIII zusammen, wobei dieses Risiko innerhalb der ersten 20 Expositionstage am höchsten ist. Die berichtete Inzidenz von inhibitorischen Antikörpern bei Patienten mit schwerer Hämophilie A und hohem Risiko für eine Inhibitorentwicklung (z. B. bislang unbehandelte Patienten) wird in Studien für Recombinate Antihämophilie Faktor mit 31 % angegeben. Das liegt innerhalb des für plasmatischen AHF berichteten Bereiches. Patienten, die mit Recombinate Antihämophilie Faktor (rekombinant) 500 behandelt werden, sollten sorgfältig mittels klinischer Überwachung und durch Labortests auf die Entwicklung von Inhibitoren hin überprüft werden.
Kinder und Jugendliche
Mit Ausnahme der Inhibitorbildung bei zuvor unbehandelten pädiatrischen Patienten (PUPs), wurden in klinischen Studien keine alters-spezifischen Unterschiede bei den Nebenwirkungen beobachtet.
4.9 Überdosierung
Symptome durch Überdosierung sind nicht bekannt.
5. PHARMAKOLOGISCHE EIGENSCHAFTEN
5.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Antihämorrhagika: Blutgerinnungsfaktor VIII, ATC-Code B02BD02.
Der Faktor VIII/von-Willebrand-Faktor-Komplex besteht aus zwei Molekülen (Faktor VIII und von-Willebrand-Faktor) mit verschiedenen physiologischen Funktionen.
Wenn er einem hämophilen Patienten infundiert wird, bindet sich der Faktor VIII an den von-Willebrand-Faktor im Blutkreislauf des Patienten.
Aktivierter Faktor VIII wirkt als Kofaktor für aktivierten Faktor IX und beschleunigt die Umwandlung von Faktor X in aktivierten Faktor X. Der aktivierte Faktor X wandelt Prothrombin in Thrombin um. Thrombin wandelt dann Fibrinogen in Fibrin um und ein Gerinnsel kann sich bilden. Die Hämophilie A ist eine geschlechtsgebundene Erbkrankheit, die eine zu geringe Konzentration an Faktor VIII:C bedingt. Sie resultiert in starken Blutungen in Gelenke, Muskeln oder innere Organe, die spontan oder als Folge von Unfällen oder chirurgischen Traumata auftreten können. Die Substitutionstherapie hebt den Plasmaspiegel des Faktor VIII an und ermöglicht so eine vorübergehende Korrektur des Faktor VIII-Mangels und der Blutungsneigung.
Recombinate Antihämophilie Faktor wurde in einer klinischen Studie bei 71 vorher unbehandelten Kindern (PUPs) untersucht. Das mittlere Alter der Gruppe bei der ersten Recombinate Antihämophilie Faktor (rekombinant)-Infusion lag bei 10 Monaten (zwischen 2 Tagen und 50 Monaten). Das Produkt wurde gut vertragen und zeigte keine signifikanten Kurzzeit-Nebenwirkungen. Seine klinische Wirksamkeit war sowohl hinsichtlich der akuten Blutstillung als auch bei der operativen Prophylaxe (10 Patienten mussten sich Operationen unterziehen) vergleichbar mit anderen Faktor VIII-Molekülen, die die volle Länge aufwiesen. Eine Langzeitbeobachtung der Gruppe zeigte eine produktbezogene Nebenwirkungsrate von 0,86 pro 1000 Infusionen. Es wurden keine schwerwiegenden oder lebensbedrohlichen Ereignisse beobachtet.
5.2 Pharmakokinetische Eigenschaften
Bei pharmakokinetischen Untersuchungen an 69 zuvor behandelten Patienten (PTPs) wurde eine mittlere Halbwertszeit für Recombinate Antihämophilie Faktor (rekombinant) 500 im Blutkreislauf von 14,6 ± 4,9 Stunden ermittelt (n=67), was statistisch keinen signifikanten Unterschied zum plasmatischen Anithämophi-liefaktor Hemofil M (plasm. AHF) darstellt. Die mittlere Halbwertszeit von Hemofil M betrug 14,7 ± 5,1 Stunden (n=61). Beim Vergleich mit dem aus Plasma gewonnenen (humanen) Faktor VIII, nach der Infusion einer Dosis von 50 I.E./kg Recombinate Antihämophilie Faktor, wurde eine tatsächliche Recovery von 123,9 ± 47,7 I.E./dl festgestellt (n=23), was signifikant höher als die tatsächliche Recovery von Hemofil M mit 101,7 ± 31,6 I.E./dl ist. Jedoch ist das Verhältnis der errechneten zu der tatsachlichen Recovery (z. B. 2 % Anstieg der Faktor VIII-Aktivität pro 1 I.E. rekomb. AHF/kg Körpergewicht) bei Recombinate Antihämophilie Faktor (121,2 ± 48,9 %) ähnlich dem bei Hemofil M ermittelten Verhältnis (123,4 ± 16,4 %).
Mit 68 zuvor unbehandelten Patienten wurden 494 Recovery-Tests durchgeführt. 212 Recovery-Tests wurden mit Patienten durchgeführt, die wegen Blutungen behandelt wurden, wobei eine mittlere tatsächliche Recovery ± SD von 70,0 ± 37,9 I.E./dl (n=208) festgestellt wurde (vier Recovery-Tests wurden als "Ausreißer" nicht in die Analyse eingeschlossen). Die hohe Abweichung ergibt sich aus den unterschiedlichen Dosierungen, die zwischen 13,8 und 103,2 I.E./kg (Mittelwert ± SD: 36,0 ± 16,2 und Median: 30,2 I.E./kg). Um die unterschiedlichen Dosierungen ebenfalls zu berücksichtigen, wurde das Verhältnis zwischen tatsächlicher und erwarteter Recovery berechnet, wobei sich ein Mittelwert von 1,0 ± 0,3 ergab.
Insgesamt wurden 68 Recovery-Tests bei Patienten durchgeführt, die im Rahmen einer weiterführenden Behandlung von vorhergehenden Blutungen Infusionen erhielten. Der tatsächliche Faktor VIII-Recovery-Wert wurde dabei auf den Wert vor der Infusion korrigiert. Die mittlere tatsächliche Recovery ± SD lag bei 88,6 ± 38,2 I.E./dl (n= 66) (zwei Recovery-Tests wurden als "Ausreißer" nicht in die Analyse eingeschlossen). Auch in diesem Fall ergaben sich durch die unterschiedlichen Dosierungen von 18,5 bis 85,7 I.E./kg (Mittelwert ± SD: 38,6 ± 15,9 und Median: 32,1 I.E./kg) große Unterschiede in den beobachteten Recovery-
Werten. Das mittlere Verhältnis zwischen tatsächlicher und erwarteter Recovery ± SD lag bei 1,0 ± 0,3 bei einem Median von 1,0.
Insgesamt 214 Recovery-Tests wurden bei Patienten in stabilem Zustand durchgeführt, wobei sich eine mittlere tatsächliche Recovery von 71,6 ± 29,7 I.E./dl (n= 209) ergab (fünf Recovery-Tests wurden als "Ausreißer" nicht in die Analyse eingeschlossen). Es wurden Dosierungen zwischen 10,4 und 68,1 I.E./kg verabreicht (Mittelwert ± SD: 38,0 ± 12,7 und Median: 36,1 I.E./kg). Das mittlere Verhältnis zwischen tatsächlicher und erwarteter Recovery ± SD lag bei 1,0 ± 0,3.
5.3 Präklinische Daten zur Sicherheit
Recombinate Antihämophilie Faktor (rekombinant) 500 verhält sich wie der endogene Faktor VIII. Dosen, die die empfohlene Dosierung zur Anwendung am Menschen pro kg Körpergewicht um ein Mehrfaches überschreiten, zeigen bei Labortieren keine toxischen Effekte. Recombinate Antihämophilie Faktor wurde in Dosen, die die Plasmakonzentration von Faktor VIII beträchtlich überschritten in vitro, und in Dosen bis zum Zehnfachen der erwarteten klinischen Höchstdosis in vivo, auf Mutagenität untersucht. Es verursachte keine Rückmutationen, Chromosomenschäden oder einen Anstieg der Mikronuklei in den polychromatischen Erythrozyten des Knochenmarks. Da die klinischen Erfahrungen keine Hinweise auf tumorigene und mutagene Effekte ergaben, werden Langzeitstudien an Tieren zur Evaluierung des kanzerogenen Potentials als nicht zwingend erachtet.
6. PHARMAZEUTISCHE ANGABEN
6.1 Liste der sonstigen Bestandteile
Pulver:
Albumin vom Menschen
Natriumchlorid
Histidin
Macrogol 3350 Kalziumchlorid-Dihydrat
Lösungsmittel:
Wasser für Injektionszwecke
6.2 Inkompatibilitäten
Da Kompatibilitätsstudien fehlen, darf dieses Arzneimittel nicht mit anderen Arzneimitteln oder Lösungen gemischt werden.
Es dürfen nur die mitgelieferten Infusionssets verwendet werden, da Therapieversagen als Folge einer Adsorption von humanem Gerinnungsfaktor VIII an der inneren Oberfläche mancher Infusionssets auftreten kann.
6.3 Dauer der Haltbarkeit
2 Jahre. Nach der Rekonstitution darf das Produkt nicht wieder gekühlt werden und sollte innerhalb von drei Stunden verbraucht werden.
6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Im Kühlschrank lagern (bei 2 °C bis 8 °C).
Nicht einfrieren.
Im Umkarton aufbewahren, um das Produkt vor Licht zu schützen.
Innerhalb der Laufzeit darf das Produkt bis zu sechs Monaten bei 15 °C bis 25 °C gelagert werden.
Nach Lagerung bei Raumtemperatur (15 °C bis 25 °C) nicht wieder kühlen.
Zu den Lagerungsbedingungen des rekonstituierten Arzneimittels siehe Abschnitt 6.3.
6.5 Art und Inhalt des Behältnisses
Eine Einzelpackung enthält eine Durchstechflasche mit Pulver, eine Durchstechflasche mit 10 ml Lösungsmittel (beide Glas Typ I mit Gummistopfen) und ein Medizinprodukt zur Rekonstitution (BAXJECT II) + 1 sterile KunststoffEinmalspritze + 1 steriles Mini-Infusionsset + 2 Alkoholtupfer + 2 Pflaster.
Alternativ zum BAXJECT II kann für Recombinate Antihämophilie Faktor (rekombinant) 500 ein Medizinprodukt zur Rekonstitution zur Verfügung gestellt werden, bestehend aus einer sterilen, doppelseitigen Nadel (zum Transfer des Lösungsmittels in die Recombinate Antihämophilie Faktor (rekombinant) 500-Flasche) und einer sterilen Filternadel (zum Transfer der rekonstituierten Lösung in die Spritze)
Packungsgröße: 1 Stück
6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung
Das Präparat wird nach Auflösen mit dem mitgelieferten, sterilisierten Wasser für Injektionszwecke intravenös verabreicht. Hierfür sollte die mitgelieferte Einmalspritze aus Kunststoff verwendet werden.
- Das Präparat innerhalb von drei Stunden nach dem Auflösen verwenden.
- Das Produkt nach dem Auflösen nicht mehr kühlen.
Nicht verwendetes Produkt oder Abfallmaterial gemäß den lokalen Vorschriften entsorgen.
Die Lösung soll klar oder leicht opaleszent sein. Trübe Lösungen oder solche mit Niederschlag dürfen nicht verwendet werden. Rekonstituiertes Produkt vor Verabreichung visuell auf Schwebeteilchen oder Verfärbung überprüfen.
Nicht verwenden, wenn das Produkt, seine sterile Barriere oder seine Verpackung beschädigt ist oder Zeichen von Veränderungen aufweist.
AUFLÖSEN: AUF ASEPTISCHE ARBEITSWEISE ACHTEN
|
Rekonstitution mit dem BAXJECT II |
Rekonstitution mit Nadeln |
|
1. Recombinate Antihämophilie Faktor (rekombinant) 500 (Pulver) und das sterilisierte Wasser für Injektionszwecke (Lösungsmittel) auf 15 °C bis 25 °C bringen. |
1. Recombinate Antihämophilie Faktor (rekombinant) 500 (Pulver) und das sterilisierte Wasser für Injektionszwecke (Lösungsmittel) auf 15 °C bis 25 °C bringen. |
|
2. Schutzkappen von den Pulver- und Lösungsmittelflaschen entfernen. |
2. Schutzkappen von den Pulver- und Lösungsmittelflaschen entfernen. |
|
3. Gummistopfen mit Alkoholtupfer reinigen. Stellen Sie die Durchstechflaschen auf eine gerade Oberfläche. |
3. Gummistopfen mit Alkoholtupfer reinigen. Stellen Sie die Durchstechflaschen auf eine gerade Oberfläche. |
|
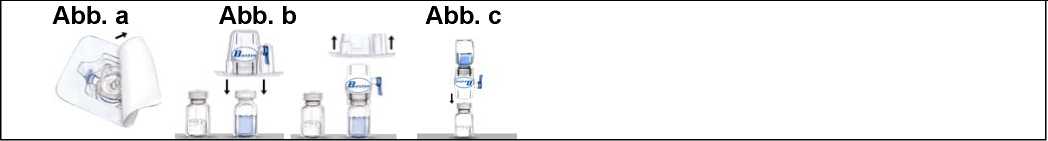
4. Die Verpackung des BAXJECTs II öffnen, indem die Schutzfolie abgezogen wird, ohne dabei den Packungsinhalt zu berühren (Abb. a). Den BAXJECT II noch nicht aus der Verpackung nehmen. |
4. Die Schutzkappen von einem Ende der doppelseitigen Nadel entfernen und dieses Ende der Nadel durch den Gummistopfen stechen. |
|
5. Die Packung nach unten drehen und den durchsichtigen Plastikdorn durch den Gummistopfen der Lösungsmittelflasche drücken. Fassen Sie die Verpackung am Rand und ziehen Sie den BAXJECT II heraus (Abb. b). Noch nicht die blaue Kappe vom BAXJECT II entfernen. |
5. Die Schutzkappe vom anderen Ende der doppelseitigen Nadel entfernen und das Lösungsmittel kopfüber über die Recombinate Antihämophilie Faktor (rekombinant) 500-Flasche halten. Dann schnell das freie Ende der Nadel durch den Stopfen der Recombinate Antihämophilie Faktor (rekombinant) 500-Flasche stechen. Das Vakuum wird das Lösungsmittel in die Recombinate Antihämophilie Faktor (rekombinant) 500-Flasche ziehen. |
|
6. Das System, bestehend aus dem BAXjEct II und der daran fixierten Lösungsmittelflasche, nun so wenden, dass sich die Lösungsmittelflasche oberhalb des BAXJECT II befindet. Den weißen Plastikdorn durch den Stopfen der Recombinate Antihämophilie Faktor (rekombinant) 500-Flasche stechen. Das |
6. Beide Durchstechflaschen trennen, indem die Nadel aus dem Gummistopfen der Lösungsmittel-Flasche gezogen wird. Dann die Nadel von der Recombi-nate Antihämophilie Faktor (rekombinant) 500-Flasche entfernen. So lange vorsichtig schwenken, bis das ganze Material gelöst ist. Sicherstellen, dass |
|
16 |
Seite 13 von |
Vakuum wird das Lösungsmittel in die Recombinate Antihämophilie Faktor (rekombinant) 500-Flasche ziehen (Abb. c).
Recombinate Antihämophilie Faktor (rekombinant) 500 vollständig aufgelöst ist, da sonst Wirksubstanz durch die Filternadel zurückgehalten wird._
7. So lange vorsichtig schwenken, bis das ganze Material gelöst ist. Sicherstellen, dass Recombinate Antihämophilie Faktor (rekombinant) 500 vollständig aufgelöst ist, da sonst Wirksubstanz durch den im Medizinprodukt enthaltenen Filter zurückgehalten wird. Das Produkt löst sich schnell auf (normalerweise innerhalb einer Minute).
|
VERABREICHUNG: AUF ASEPTISCHE ARBEITSWEISE ACHTEN | |
|
Es wird empfohlen, die Verabreichung innerhalb von drei Stunden nach dem Auflösen zu beginnen und Recombinate Antihämophilie Faktor (rekombinant) 500 im aufgelösten Zustand nicht zu kühlen. Rekonstituiertes Produkt vor der Anwendung auf Partikel und Verfärbung prüfen. Recombinate Antihämophilie Faktor (rekombinant) 500 kann farblos bis leicht gelblich sein. 1. Die blaue Schutzkappe vom BAXJECT II abziehen. KEINE LUFT IN DIE FERTIGSPRITZE AUFZIEHEN. Die Spritze mit dem BAXJECT II verbinden (Abb. d). |
Es wird empfohlen, die Verabreichung innerhalb von drei Stunden nach dem Auflösen zu beginnen und Recombinate Antihämophilie Faktor (rekombinant) 500 im aufgelösten Zustand nicht zu kühlen. Rekonstituiertes Produkt vor der Anwendung auf Partikel und Verfärbung prüfen. Recombinate Antihämophilie Faktor (rekombinant) 500 kann farblos bis leicht gelblich sein. 1. Die Filternadel an die Fertigspritze anschließen und den Kolben zurückziehen, um Luft in die Spritze aufzuziehen. |
|
2. Das System umdrehen (so dass sich die Produkt-Flasche oben befindet). Durch langsames Zurückziehen des Kolbens das Konzentrat in die Spritze aufziehen (Abb. e.) |
2. Die Filternadel in das rekonstituierte Recombinate Antihämophilie Faktor (rekombinant) 500 einstechen. |
|
3. Die Spritze entfernen. |
3. Luft in die Durchstechflasche injizieren und dann das rekonstituierte Material in die Spritze aufziehen. |
|
4. Das Infusionsset an die Spritze anschließen. Langsam intravenös injizieren. Die Zubereitung kann mit einer Infusionsgeschwindigkeit von bis zu 10 ml pro Minute verabreicht werden. Die Pulsfrequenz des Patienten sollte vor und während der Verabreichung von Recombinate |
4. Die Filternadel abziehen und verwerfen. Das Infusionsset an die Spritze anschließen. Langsam intravenös injizieren. Die Zubereitung kann mit einer Infusionsgeschwindigkeit von bis zu 10 ml pro Minute verabreicht werden. Die Pulsfrequenz des Patienten sollte vor und während der Verabreichung von |
|
Antihämophilie Faktor (rekombinant) 500 gemessen werden. Eine deutliche Erhöhung der Pulsfrequenz kann durch Verlangsamen oder zeitweiliges Unterbrechen der Injektion meist sofort wieder gesenkt werden (siehe Abschnitte 4.4 und 4.8). |
Recombinate Antihämophilie Faktor (rekombinant) 500 gemessen werden. Eine deutliche Erhöhung der Pulsfrequenz kann durch Verlangsamen oder zeitweiliges Unterbrechen der Injektion meist sofort wieder gesenkt werden (siehe Abschnitte 4.4 und 4.8). |
|
Abb. d Abb. e * <’ w - m' ^ B |
5. Zur Entnahme des rekonstituierten Recombinate Antihämophilie Faktor (rekombinant) 500 muss für jede weitere Durchstechflasche eine neue Filternadel benutzt werden. |
7. INHABER DER ZULASSUNG
Baxter Deutschland GmbH Edisonstraße 4 85716 Unterschleißheim Telefon: 089/31701-0 Fax: 089/31701-177 E-Mail: info de@baxter.com
8. ZULASSUNGSNUMMER
28530.01.00
9. DATUM DER ERTEILUNG DER ZULASSUNG/VERLÄNGERUNG DER ZULASSUNG
Datum der Zulassung: 27. Juli 1993
Datum der Verlängerung der Zulassung: 11. Mai 2003
10. STAND DER INFORMATION
Februar 2011
11. VERKAUFSABGRENZUNG
Verschreibungspflichtig
12. STOFF- ODER INDIKATIONSGRUPPE
Anthämorrhagikum rekombinant
13. SONSTIGE HINWEISE
Herkunftsländer der zur Produktion verwendeten Plasmen:
Deutschland, Norwegen, Österreich, Schweiz, Schweden, Tschechien und Vereinigte Staaten von Amerika.
Seite 16 von
16