Rolenium 50 Mikrogramm /250 Mikrogramm /Dosis, Einzeldosiertes Pulver Zur Inhalation
Fachinformation
1. Bezeichnung des Arzneimittels
Rolenium® 50 Mikrogramm/250 Mikrogramm/Dosis, einzeldosiertes Pulver zur Inhalation
Rolenium® 50 Mikrogramm/500 Mikrogramm/Dosis, einzeldosiertes Pulver zur Inhalation
2. Qualitative und quantitative Zusammensetzung
Rolenium® 50 Mikrogramm/250 Mikrogramm/Dosis, einzeldosiertes Pulver zur Inhalation
Jede Einzeldosis Rolenium® enthält
50 Mikrogramm Salmeterol (als Salmeterolxinafoat) und 250 Mikrogramm Fluticasonpropionat.
Sonstiger Bestandteil mit bekannter Wirkung:
Enthält 24,677 mg Lactose-Monohydrat.
Rolenium® 50 Mikrogramm/500 Mikrogramm/Dosis, einzeldosiertes Pulver zur Inhalation
Jede Einzeldosis Rolenium® enthält
50 Mikrogramm Salmeterol (als Salmeterolxinafoat) und 500 Mikrogramm Fluticasonpropionat.
Sonstiger Bestandteil mit bekannter Wirkung:
Enthält 24,427 mg Lactose-Monohydrat.
Vollständige Auflistung der sonstigen Bestandteile siehe Abschnitt 6.1.
3. Darreichungsform
Einzeldosiertes Pulver zur Inhalation.
4. Klinische Angaben
4.1 Anwendungsgebiete
Asthma
Rolenium® wird als Standardbehandlung von Asthma angewendet, bei der die Anwendung eines Kombinationspräparats (langwirkender Beta-2-Agonist und inhalative Corticosteroide) angebracht ist:
- Patienten, die mit inhalativen Corticosteroiden nicht ausreichend kontrolliert werden und die bei Bedarf kurzwirkende inhalative Beta-2-Agonisten anwenden
- Patienten, die bereits sowohl mit inhalativen Corticosteroiden als auch mit langwirkenden Beta-2-Agonisten ausreichend kontrolliert werden.
Chronisch-obstruktive Lungenerkrankung (COPD)
Rolenium® wird bei der symptomatischen Behandlung von Patienten mit COPD angewendet, die eine FEV1 < 60 % des vorhergesagten Normwerts (vor Anwendung eines Bronchodilatators) und wiederholt Exazerbationen aufweisen und trotz kontinuierlicher Therapie mit Bronchodilatatoren an signifikanten Symptomen leiden.
4.2 Dosierung und Dauer der Anwendung
Rolenium® ist ausschließlich zur Inhalation bestimmt.
Patienten sollten darauf hingewiesen werden, dass es für den optimalen Behandlungserfolg erforderlich ist, Rolenium® täglich anzuwenden, auch wenn sie symptomfrei sind.
Patienten sollten regelmäßig von einem Arzt untersucht werden, um eine optimale Konzentration von Rolenium® zu erhalten und diese nur auf medizinischen Rat zu ändern. Die Dosis sollte so titriert werden, dass eine effektive Kontrolle der Symptome mit der niedrigsten Dosierung erreicht wird. In den Fällen, in denen mit der niedrigsten zweimal täglich verabreichten Konzentration der Kombination eine Kontrolle der Symptome erreicht wird, kann im nächsten Schritt eine alleinige Behandlung mit inhalativen Corticosteroiden in Betracht gezogen werden. Alternativ können Patienten, die mit einem langwirkenden Beta-2-Agonisten behandelt werden müssen, auf Rolenium® einmal täglich titriert werden, wenn es nach Meinung des verordnenden Arztes zur Kontrolle der Erkrankung angebracht scheint. Bei einer einmal täglichen Verabreichung sollte die Dosis abends verabreicht werden, wenn der Patient in der Anamnese nächtliche Symptome aufweist. Weist der Patient in der Anamnese überwiegend tagsüber Symptome auf, sollte die Dosis morgens verabreicht werden.
Patienten sollten Rolenium® in der Konzentration erhalten, die eine dem Schweregrad der Erkrankung angemessene Dosis Fluticasonpropionat enthält. Der verordnende Arzt sollte berücksichtigen, dass etwa die halbe Tagesdosis Fluticasonpropionat (in Mikrogramm) bei Patienten mit Asthma ebenso wirksam ist wie die ganze Tagesdosis anderer inhalativer Steroide. Benötigt ein Patient Dosierungen, die außerhalb der empfohlenen Dosis liegen, sollten geeignete Dosen von Beta-Agonisten und/oder Corticosteroiden verschrieben werden.
Empfohlene Dosis:
Asthma
Erwachsene:
Eine Inhalation von 50 Mikrogramm Salmeterol und 250 Mikrogramm Fluticasonpropionat zweimal täglich eine Inhalation von 50 Mikrogramm Salmeterol und 500 Mikrogramm Fluticasonpropionat zweimal täglich.
Eine kurzzeitige Probebehandlung mit Rolenium® kann bei Erwachsenen mit mittelschwerem persistierendem Asthma (definiert als: Patienten mit täglich auftretenden Symptomen, täglicher Anwendung eines Notfallmedikaments und mittelschwerer bis schwerer Einschränkung des Atemflusses), für die eine schnelle Asthmakontrolle notwendig ist, als initiale Erhaltungstherapie in Betracht gezogen werden. Bei diesen Fällen wird eine Inhalation von 50 Mikrogramm Salmeterol und 100 Mikrogramm Fluticasonpropionat zweimal täglich als Anfangsdosis empfohlen. Sobald die Kontrolle des Asthmas erreicht wurde, sollte die Behandlung überprüft und eine stufenweise Reduzierung der Therapie auf eine alleinige Behandlung mit inhalativen Corticosteroiden in Betracht gezogen werden. Während der stufenweisen Reduzierung der Behandlung ist eine regelmäßige Untersuchung des Patienten erforderlich.
Im Vergleich zu einer initialen Erhaltungstherapie mit inhalativem Fluticasonpropionat zeigte sich kein deutlicher Vorteil, wenn ein oder zwei Kriterien des Schweregrads nicht erfüllt waren. Allgemein bleiben inhalative Corticosteroide für die meisten Patienten die First-Line-Therapie. Rolenium® ist nicht zur Initialbehandlung von leichtem Asthma vorgesehen. Salmeterol/Fluticasonpropionat ist in der Stärke 50 Mikrogramm/100 Mikrogramm nicht für Erwachsene mit schwerem Asthma geeignet. Bevor eine fixe Kombinationsdosis bei Patienten mit schwerem Asthma angewendet werden kann, wird die Festlegung einer angemessenen Dosis inhalativer Corticosteroide empfohlen.
COPD
Erwachsene:
Eine Inhalation von 50 Mikrogramm Salmeterol und 500 Mikrogramm Fluticasonpropionat zweimal täglich.
Besondere Patientengruppen:
Bei älteren Patienten oder bei Patienten mit eingeschränkter Nierenfunktion ist keine Anpassung der Dosis erforderlich. Für die Anwendung von Salmeterol/Fluticasonpropionat bei Patienten mit eingeschränkter Leberfunktion liegen keine Daten vor.
Kinder und Jugendliche:
Rolenium® darf bei Kindern und Jugendlichen nicht angewendet werden.
4.3 Gegenanzeigen
Rolenium® darf bei Patienten mit Überempfindlichkeit (Allergie) gegen einen der Wirkstoffe oder einen der sonstigen Bestandteile von Rolenium® nicht angewendet werden (siehe Abschnitt 6.1).
4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Die Behandlung von Asthma erfolgt im Normalfall schrittweise, und das Ansprechen des Patienten auf die Behandlung sollte klinisch beobachtet und mit Lungenfunktionstests überprüft werden.
Rolenium® sollte nicht zur Behandlung akuter Asthmasymptome angewendet werden, für die schnell- und kurzwirkende Bronchodilatatoren notwendig sind. Patienten sollte empfohlen werden, ihr Arzneimittel für die Behandlung eines akuten Asthmaanfalls stets mit sich zu führen.
Eine Behandlung mit Rolenium® sollte nicht während einer Exazerbation oder bei sich signifikant verschlechterndem oder akut verschlimmerndem Asthma begonnen werden.
Während der Behandlung mit Rolenium® können schwerwiegende mit Asthma einhergehende unerwünschte Ereignisse und Exazerbationen auftreten. Patienten sollten gebeten werden, die Behandlung fortzusetzen, jedoch ärztlichen Rat einzuholen, wenn die Asthmasymptome unkontrolliert bleiben oder sich nach dem Beginn einer Therapie mit Rolenium® verschlechtern.
Eine erhöhte Anwendung kurzwirkender Bronchodilatatoren zur Linderung der Symptome weist auf eine verschlechterte Kontrolle hin. Die Patienten sollten dann von einem Arzt untersucht werden.
Eine plötzliche und fortschreitende Verschlechterung der Asthmakontrolle kann möglicherweise lebensgefährlich sein und der Patient sollte sich umgehend einer medizinischen Untersuchung unterziehen. Eine Steigerung der Corticosteroid-Dosis sollte in Betracht gezogen werden. Patienten, bei denen die aktuelle Dosierung Rolenium® keine angemessene Asthmakontrolle ermöglicht, sollten medizinisch untersucht werden.
Sobald die Asthmasymptome unter Kontrolle gebracht wurden, kann eine Reduzierung der Dosis Rolenium® in Betracht gezogen werden. Während der schrittweisen Reduzierung der Behandlung ist eine regelmäßige Untersuchung des Patienten erforderlich. Die niedrigste wirksame Dosis Rolenium® sollte angewendet werden (siehe Abschnitt 4.2).
Bei Patienten mit Asthma oder COPD sollte eine zusätzliche Therapie mit Corticosteroiden in Betracht gezogen werden.
Die Behandlung mit Rolenium® sollte bei Patienten mit Asthma nicht aufgrund des Risikos einer Exazerbation abrupt abgebrochen werden. Die Therapie sollte unter Aufsicht eines Arztes heruntertitriert werden. Bei Patienten mit COPD kann ein Abbruch der Therapie mit einer symptomatischen Dekompensation einhergehen und sollte unter Aufsicht eines Arztes erfolgen.
Wie alle inhalativen Präparate, die Corticosteroide enthalten, sollte Rolenium® bei Patienten mit pulmonaler T uberkulose mit Vorsicht angewendet werden.
Hohe Dosen Rolenium® können in seltenen Fällen Herzrhythmusstörungen wie z.B. supraventrikuläre Tachykardie, Extrasystolen und Vorhofflimmern sowie eine leichte vorübergehende Senkung der Kaliumkonzentration im Serum verursachen. Deshalb sollte Rolenium® bei Patienten mit schweren kardiovaskulären Erkrankungen, Herzrhythmusstörungen, Diabetes mellitus, Thyreotoxikose, nicht eingestellter Hypokaliämie oder Patienten mit der Neigung zu einer niedrigen Kaliumkonzentration im Serum mit Vorsicht angewendet werden.
In sehr seltenen Fällen wurde von einem Anstieg des Glucosegehalts im Blut berichtet (siehe Abschnitt 4.8). Dies sollte bei der Verordnung des Medikaments für Patienten mit Diabetes mellitus in der Anamnese berücksichtigt werden.
Wie bei anderen Inhalationstherapien können nach Verabreichung paradoxe Bronchospasmen mit sofort einsetzender erhöhter Stenoseatmung auftreten. Rolenium® sollte umgehend abgesetzt, der Patient untersucht und gegebenenfalls eine Alternativbehandlung begonnen werden.
Rolenium® enthält Lactose. Patienten mit der seltenen hereditären Galactose-Intoleranz, Lactase-Mangel oder Glucose-Galactose-Malabsorption sollten dieses Arzneimittel nicht anwenden.
Patienten sollten mit Sorgfalt auf eine Therapie mit Rolenium® umgestellt werden, insbesondere wenn aufgrund der vorhergehenden systemischen Therapie mit Steroiden eine beeinträchtigte Nebennierenfunktion zu vermuten ist.
Systemische Nebenwirkungen können bei der Behandlung mit jedem inhalativen Corticosteroid auftreten, insbesondere wenn hohe Dosen über einen langen Zeitraum verordnet werden. Die Wahrscheinlichkeit des Auftretens dieser Nebenwirkungen ist weniger wahrscheinlich als bei oralen Corticosteroiden. Zu den möglichen systemischen Nebenwirkungen zählen Cushing-Syndrom, cushingoide Erscheinungen, adrenale Suppression, Wachstumsverzögerung bei Kindern und Jugendlichen, abnehmende Knochenmineraldichte, Katarakt, Glaukom und sehr selten eine Reihe von psychologischen oder verhaltensbezogenen Wirkungen wie psychomotorische Hyperaktivität, Schlafstörungen, Angstzustände, Depressionen oder aggressives Verhalten (insbesondere bei Kindern). Deshalb ist es wichtig, dass der Patient regelmäßig untersucht wird und die Dosis inhalativer Corticosteroide auf die niedrigste Dosismenge gesenkt wird, mit der eine wirksame Asthmakontrolle erreicht wird.
Werden hohe Dosen inhalativer Corticosteroide über einen längeren Zeitraum verabreicht, kann dies bei Patienten zu einer adrenalen Suppression und einer akuten Nebenniereninsuffizienz führen. In sehr seltenen Fällen wurde bei Dosen zwischen 500 und weniger als 1000 Mikrogramm Fluticasonpropionat von einer adrenalen Suppression und Nebennierenkrise berichtet. Zu den Faktoren, die eine akute Nebennierenkrise begünstigen können, zählen Traumata, Operationen, Infektionen oder eine abrupte Reduzierung der Dosismenge. Die auftretenden Symptome sind normalerweise nicht eindeutig. Dazu zählen Anorexie, Bauchschmerzen, Gewichtsverlust, Müdigkeit, Kopfschmerzen, Übelkeit, Erbrechen, Hypotonie, eine Bewusstseinseintrübung, Hypoglykämie und Krampfanfälle. In Stressphasen oder bei selektiven Operationen sollte eine zusätzliche Behandlung mit systemischen Corticosteroiden in Betracht gezogen werden.
Der Nutzen einer Therapie mit inhalativem Fluticasonpropionat sollte den Bedarf an oralen Steroiden senken. Jedoch kann bei Patienten, deren Therapie von oralen Steroiden auf Fluticasonpropionat umgestellt wird, das Risiko einer eingeschränkten Nebennierenreserve über einen langen Zeitraum bestehen bleiben. Auch bei Patienten, die in der Vergangenheit hohe Dosen Corticosteroide als Notfallmedikament benötigten, kann ein Risiko bestehen. Die Möglichkeit einer zurückbleibenden Funktionsstörung sollte in Notfällen und in elektiven Situationen, die vermutlich Stress verursachen können, stets berücksichtigt werden sowie eine angemessene Behandlung mit Corticosteroiden in Betracht gezogen werden. Je nach Ausmaß der Nebennierenfunktionsstörung muss vor elektiven Maßnahmen der Rat eines Facharztes eingeholt werden.
Ritonavir kann die Fluticasonpropionatkonzentration im Plasma erheblich steigern. Deshalb sollte eine gleichzeitige Anwendung vermieden werden, es sei denn, der Nutzen für den Patienten übersteigt die Risiken systemischer Nebenwirkungen von Corticosteroiden. Auch bei der Kombination von Fluticasonpropionat mit anderen potenten CYP3A-Hemmern besteht ein erhöhtes Risiko systemischer
Nebenwirkungen (siehe Abschnitt 4.5).
Im Rahmen der TORCH-Studie wurde bei Patienten mit COPD, die mit Salmeterol/ Fluticasonpropionat 50/500 Mikrogramm zweimal täglich behandelt wurden, im Vergleich zu Placebo sowie in den Studien SCO40043 und SCO1000250, in denen die niedrigere, nicht für Patienten mit COPD zugelassene Dosis von Salmeterol/Fluticasonepropionat 50/250 Mikrogramm zweimal täglich mit nur Salmeterol 50 Mikrogramm zweimal täglich verglichen wurde, häufiger von Infektionen der unteren Atemwege (insbesondere Pneumonie und Bronchitis) berichtet (siehe Abschnitt 4.8 und 5.1). In sämtlichen Studien wurde in der Salmeterol/Fluticasonpropionat-Gruppe eine ähnliche Häufigkeit von Pneumonien beobachtet.
In der TORCH-Studie war bei älteren Patienten, Patienten mit einem niedrigen Body-Mass-Index (< 25 kg/m2) und Patienten mit sehr schwerer Erkrankung (FEV1 < 30 % des vorhergesagten Normwerts) das Risiko einer Pneumonie, unabhängig von der Behandlung, am höchsten. Ärzte sollten bei Patienten mit COPD besonders auf das mögliche Auftreten einer Pneumonie oder einer anderen Infektion der unteren Atemwege achten, da die Symptome solcher Infektionen sich mit der einer Exazerbation häufig überschneiden. Hatte ein Patient mit schwerer COPD bereits eine Pneumonie, sollte die Behandlung mit Salmeterol/Fluticasonpropionat überdacht werden.
Daten einer großen klinischen Studie (der Salmeterol Multicenter Asthma Research Trial, SMART) ließen darauf schließen, dass bei Patienten afrikanischamerikanischer Herkunft bei der Anwendung von Salmeterol verglichen mit Placebo ein erhöhtes Risiko atemwegsbezogener schwerwiegender Ereignisse oder Todesfälle besteht (siehe Abschnitt 5.1). Unbekannt ist, ob dies von pharmakogenetischen oder anderen Faktoren abhängig war. Patienten schwarzafrikanischer oder afrokaribischer Herkunft sollten deshalb gebeten werden, die Behandlung fortzusetzen, jedoch ärztlichen Rat einzuholen, wenn die Asthmasymptome unkontrolliert bleiben oder sich während der Behandlung mit Rolenium® verschlechtern.
Bei gleichzeitiger Anwendung von systemischem Ketoconazol wird die systemische Salmeterolexposition signifikant erhöht. Dies kann zu einer erhöhten Inzidenz systemischer Nebenwirkungen führen (z.B. Verlängerung des QTc-Intervalls und Palpitationen). Eine gleichzeitige Behandlung mit Ketoconazol oder anderen potenten CYP3A4-Hemmern sollte daher vermieden werden, es sei denn, der Nutzen übersteigt das potenziell erhöhte Risiko systemischer Nebenwirkungen einer Salmeterolbehandlung (siehe Abschnitt 4.5).
Auswirkungen bei Fehlgebrauch zu Dopingzwecken
Die Anwendung von Rolenium® kann bei Dopingkontrollen zu positiven Ergebnissen führen.
4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Sowohl die Behandlung mit nicht-selektiven als auch mit selektiven Beta-Blockern sollte vermieden werden, es sei denn, es gibt ausschlaggebende Gründe für deren Anwendung.
Die gleichzeitige Anwendung anderer beta-adrenerger Arzneimittel kann potentiell additiv wirken.
Fluticasonpropionat
Unter normalen Umständen wird nach einer inhalierten Dosis Fluticasonpropionat aufgrund eines hohen First-Pass-Effekts und einer hohen systemischen Clearance durch das Cytochrom P450 3A4 in Darm und Leber eine geringe Konzentration im Plasma erreicht. Aufgrund dessen sind klinisch signifikante von Fluticasonpropionat ausgelöste Wechselwirkungen mit anderen Arzneimitteln unwahrscheinlich.
Im Rahmen einer Interaktionsstudie mit gesunden Probanden zu intranasal verabreichtem Fluticasonpropionat erhöhte Ritonavir 100 mg b.i.d. (ein
hochwirksamer Cytochrom P450 3A4-Hemmer) die Konzentrationen von Fluticasonpropionat im Plasma um mehrere hundert Mal. Dies führte zu einer deutlich reduzierten Cortisolkonzentration im Serum.
Zu Wechselwirkungen mit inhalativ verabreichtem Fluticasonpropionat liegen keine Daten vor, jedoch wird ein deutlicher Anstieg der Konzentration von Fluticasonpropionat im Plasma erwartet. Fälle von Cushing-Syndrom und adrenaler Suppression wurden berichtet. Eine Kombination sollte vermieden werden, es sei denn, der Nutzen übersteigt das erhöhte Risiko systemischer Nebenwirkungen von Glucocorticoiden.
In einer kleinen Studie mit gesunden Freiwilligen erhöhte der etwas weniger wirksame CYP3A-Hemmer Ketoconazol die Exposition von Fluticasonpropionat nach einer einzigen Inhalation um 150 %. Dies führte verglichen mit einer alleinigen Gabe von Fluticasonpropionat zu einer stärker reduzierten Cortisolkonzentration im Plasma. Bei einer Kombinationstherapie mit anderen potenten CYP3A-Hemmern wie Itraconazol wird ebenfalls eine erhöhte systemische Exposition von Fluticasonpropionat und ein Risiko systemischer Nebenwirkungen erwartet. Vorsicht ist geboten und eine Langzeitbehandlung mit diesen Arzneimitteln sollte, wenn möglich, vermieden werden.
Salmeterol
Potente CYP3A4-Hemmer
Eine Kombinationstherapie von Ketoconazol (400 mg, oral, einmal täglich) mit Salmeterol (50 Mikrogramm, inhalativ, zweimal täglich) bei 15 gesunden Probanden über einen Zeitraum von 7 Tagen führte zu einer signifikant erhöhten Plasmaexposition von Salmeterol (1,4-fache Cmax und 15-fache AUC).
Dies kann, verglichen mit einer alleinigen Behandlung mit Salmeterol oder Ketoconazol, zu einer erhöhten Inzidenz anderer systemischer Nebenwirkungen der Salmeteroltherapie (z.B. Verlängerung des QTc-Intervalls und Palpitationen) führen (siehe Abschnitt 4.4).
In Bezug auf Blutdruck, Herzfrequenz sowie Glucose- und Kaliumkonzentration im Blut wurden keine klinisch signifikanten Auswirkungen beobachtet. Die gleichzeitige Verabreichung von Ketoconazol führte weder zu einer erhöhten Ausscheidungshalbwertszeit von Salmeterol noch zu einer erhöhten Akkumulation von Salmeterol bei wiederholten Gaben.
Die gleichzeitige Verabreichung von Ketoconazol sollte vermieden werden, es sei denn, der Nutzen übersteigt das potentiell erhöhte Risiko systemischer Nebenwirkungen einer Salmeteroltherapie. Ein ähnliches Wechselwirkungsrisiko mit anderen potenten CYP3A4-Hemmern (z.B. Itraconazol, Telithromycin, Ritonavir) ist wahrscheinlich.
Moderate CYP 3A4-Hemmer
Bei 15 gesunden Freiwilligen ergab eine gleichzeitige Verabreichung von Erythromycin (500 mg, oral, dreimal täglich) und Salmeterol (50 Mikrogramm, inhalativ, zweimal täglich) über einen Zeitraum von 6 Tagen eine leicht, aber statistisch nicht signifikant erhöhte Salmeterolexposition (1,4-fache Cmax und 1,2fache AUC). Die gleichzeitige Gabe von Erythromycin war nicht mit schwerwiegenden Nebenwirkungen assoziiert.
4.6 Fertilität, Schwangerschaft und Stillzeit
Fertilität
Zu Auswirkungen beim Menschen liegen keine Daten vor. Jedoch zeigten tierexperimentelle Untersuchungen keine Auswirkungen von Salmeterol oder Fluticasonpropionat auf die Fertilität.
Schwangerschaft
Einige Daten zur Wirkung bei schwangeren Frauen (zwischen 300-1.000 Schwangerschaften) weisen weder auf eine zu Fehlbildungen führende Toxizität noch auf feto/neonatale Toxizität von Salmeterol und Fluticasonpropionat hin. Tierexperimentelle Untersuchungen zeigten eine Reproduktionstoxizität nach Verabreichung von Beta-2-Adrenorezeptor-Agonisten und Glucocorticosteroiden (siehe Abschnitt 5.3).
Schwangeren Frauen soll Rolenium® nur dann verabreicht werden, wenn der erwartete Nutzen für die Mutter höher ist als das potentielle Risiko für den Fötus.
Bei der Behandlung von schwangeren Frauen sollte die niedrigste wirksame Dosis Fluticasonpropionat angewendet werden, die eine angemessene Asthmakontrolle gewährleistet.
Stillzeit
Es ist nicht bekannt, ob Salmeterol und Fluticasonpropionat oder ihre Metaboliten in die Muttermilch übergehen.
In Studien wurde gezeigt, dass Salmeterol und Fluticasonpropionat und ihre Metaboliten bei laktierenden Ratten in die Milch übergehen.
Deshalb kann ein Risiko für Neugeborene/Kleinkinder, die gestillt werden, nicht ausgeschlossen werden. Eine Entscheidung muss darüber getroffen werden, ob das Stillen des Kindes oder die Therapie mit Rolenium® eingestellt wird. Dabei müssen der Vorteil des Stillens für das Kind und der Nutzen der Therapie für die Mutter berücksichtigt werden.
4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Zu den Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen wurden keine Studien durchgeführt.
4.8 Nebenwirkungen
Da Rolenium® sowohl Salmeterol als auch Fluticasonpropionat enthält, treten möglicherweise die bei jedem der Wirkstoffe zu erwartende Art und der zu erwartende Schweregrad von Nebenwirkungen auf. Es gibt keine Hinweise auf weitere Nebenwirkungen, die aus einer gleichzeitigen Verabreichung beider Wirkstoffe resultieren.
Mit Salmeterol/Fluticasonpropionat assoziierte Nebenwirkungen sind im Folgenden nach Systemorganklasse und Häufigkeit aufgeführt. Häufigkeiten sind wie folgt definiert: sehr häufig (> 1/10), häufig (> 1/100 bis < 1/10), gelegentlich (> 1/1000 bis < 1/100), selten (> 1/10.000 bis < 1/1.000), sehr selten (< 1/10.000) und nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar). Die Häufigkeiten wurden von den Daten klinischer Studien abgeleitet. Die Inzidenz bei Placebo wurde nicht berücksichtigt.
|
Systemorganklasse |
Nebenwirkung |
Häufigkeit |
|
Infektionen und parasitäre Erkrankungen |
Candidose des Mund- und Rachenraums Pneumonie Bronchitis |
Häufig Häufig1, 3 5 Häufig1,3 |
|
Erkrankungen des Immunsystems |
Überempfindlichkeitsreaktionen mit folgenden Symptomen: Überempfindlichkeitsreaktionen der Haut Angioödeme (hauptsächlich des Gesichts und des Mund- und Rachenraums) Respiratorische Symptome (Dyspnoe) Respiratorische Symptome (Bronchospasmen) Anaphylaktische Reaktionen, einschließlich anaphylaktischem Schock |
Selten Selten Gelegentlich Selten Selten |
|
Endokrine Erkrankungen |
Cushing-Syndrom, cushingoide Erscheinungen, adrenale Suppression, Wachstumsverzögerungen bei Kindern und Jugendlichen, verminderte Knochenmineraldichte |
Selten4 |
|
Stoffwechsel- und Ernährungsstörungen |
Hypokaliämie Hyperglykämie |
Häufig3 Selten4 |
|
Psychiatrische Erkrankungen |
Angstzustände Schlafstörungen und Verhaltensänderungen einschließlich psychomotorischer Hyperaktivität und Reizbarkeit (überwiegend bei Kindern) |
Gelegentlich Selten |
|
Depressionen, aggressives Verhalten (überwiegend bei Kindern) |
Nicht bekannt | |
|
Erkrankungen des Nervensystems |
Kopfschmerzen Tremor |
Sehr häufig1 Gelegentlich |
|
Augenerkrankungen |
Katarakt, Glaukom |
Selten4 |
|
Herzerkrankungen |
Palpitationen Tachykardie Kardiale Arrhythmien (einschließlich Vorhofflimmern, supraventrikuläre Tachykardie und Extrasystolen) Angina pectoris |
Gelegentlich Gelegentlich Selten Gelegentlich |
|
Erkrankungen der Atemwege, des Brustraums und des Mediastinums |
Nasopharyngitis Halsbeschwerden Heiserkeit/Dysphonie Sinusitis Paradoxe Bronchospasmen |
Sehr häufig2, 3 Gelegentlich Häufig Häufig1,3 Selten4 |
|
Erkrankungen der Haut und des |
Kontusionen |
Häufig1'3 |
|
Unterhautzellgewebes | ||
|
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen |
Muskelkrämpfe Traumatische Frakturen Arthralgie Myalgie |
Gelegentlich Häufig1,3 Häufig Häufig |
1Häufig bei Placebo berichtet 2Sehr häufig bei Placebo berichtet
3In einer Studie zu COPD über einen Zeitraum von 3 Jahren berichtet 4Siehe Abschnitt 4.4 5Siehe Abschnitt 5.1
Beschreibung ausgewählter Nebenwirkungen
Pharmakologische Nebenwirkungen einer Behandlung mit Beta-2-Agonisten wie Tremor, Palpitationen und Kopfschmerzen wurden berichtet, scheinen jedoch vorübergehend zu sein und bei regelmäßiger Behandlung zurückzugehen.
Aufgrund des Wirkstoffs Fluticasonpropionat können bei einigen Patienten Heiserkeit und Candidose (Soor) des Mund- und Rachenraums auftreten. Sowohl Heiserkeit als auch das Auftreten von Candidose können gelindert werden, wenn nach Anwendung des Produkts mit Wasser gegurgelt wird. Symptomatische Candidose kann mit einem topischen Antimykotikum behandelt werden, während dessen die Behandlung mit Rolenium® fortgesetzt werden kann.
Zu den möglichen systemischen Nebenwirkungen zählen Cushing-Syndrom, cushingoide Erscheinungen, adrenale Suppression, Wachstumsverzögerungen bei Kindern und Jugendlichen, verminderte Knochenmineraldichte, Katarakt und Glaukom (siehe Abschnitt 4.4).
4.9 Überdosierung
Aus klinischen Studien liegen keine Daten zur Überdosierung mit Rolenium®vor. Jedoch sind im Folgenden Daten zur Überdosierung mit den einzelnen Wirkstoffen aufgeführt:
Anzeichen und Symptome einer Überdosierung mit Salmeterol sind Tremor, Kopfschmerzen und Tachykardie. Gegenmittel erster Wahl sind kardioselektive Beta-Blocker, die bei Patienten mit Bronchospasmen in der Anamnese mit Vorsicht angewendet werden müssen. Sollte die Behandlung mit Rolenium® aufgrund einer Überdosierung mit dem Beta-Agonisten, der Bestandteil dieses Präparats ist, abgebrochen werden, muss eine angemessene Ersatztherapie mit Steroiden in Betracht gezogen werden. Außerdem kann eine Hypokaliämie auftreten und eine Kaliumsubstitution sollte erwogen werden.
Akut:
Eine akute Inhalation von Dosen Fluticasonpropionat, die die empfohlenen Dosen übersteigen, kann zu einer vorübergehenden adrenalen Suppression führen. Dabei muss keine Notfallmaßnahme eingeleitet werden, da die Nebennierenfunktion innerhalb einiger Tage wiederhergestellt ist, wie mit Messungen der Cortisolkonzentration im Plasma bestätigt werden kann.
Chronische Überdosierung mit inhalativem Fluticasonpropionat:
Siehe Abschnitt 4.4: Risiko einer adrenalen Suppression: Die Überwachung der Nebennierenreserve kann erforderlich sein. In Fällen von Überdosierung mit Fluticasonpropionat kann die Behandlung mit Rolenium® mit einer geeigneten Dosismenge zur Symptomkontrolle fortgesetzt werden.
5. Pharmakologische Eigenschaften
5.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Sympathomimetika und andere Mittel bei obstruktiven Atemwegserkrankungen.
ATC-Code: R03AK06
Klinische Studien mit Salmeterol/Fluticasonpropionat bei Patienten mit Asthma
In einer zwölfmonatigen Studie (Gaining Optimal Asthma ControL, GOAL) mit 3416 Patienten im Erwachsenen- und Jugendalter mit persistierendem Asthma wurde die Sicherheit und Wirksamkeit von Salmeterol/Fluticasonpropionat verglichen mit einer alleinigen Therapie mit dem inhalativen Corticosteroid (ICS) Fluticasonpropionat untersucht, um zu beurteilen, ob die Ziele der Asthmabehandlung erreicht werden können. Die Dosis wurde stufenweise alle
12 Wochen erhöht bis **eine vollständige Kontrolle oder die höchste Dosismenge des Prüfpräparats erreicht wurde. Die GOAL-Studie zeigte, dass bei mehr Patienten, die mit Salmeterol/Fluticasonpropionat behandelt wurden, das Asthma kontrolliert werden konnte als bei Patienten, die nur mit ICS behandelt wurden, und dass diese Kontrolle mit einer niedrigeren Dosis des Corticosteroids erreicht wurde.
Gut kontrolliertes Asthma wurde mit Salmeterol/Fluticasonpropionat schneller erreicht als nur mit ICS. Die Behandlungsdauer zum Erreichen einer ersten individuell gut kontrollierten Woche betrug bei 50% der Studienteilnehmer 16 Tage in der Salmeterol/Fluticasonpropionat-Gruppe, verglichen mit 37 Tagen in der ICS-Gruppe. In der Untergruppe mit steroid-naiven Asthmatikern betrug die Behandlungsdauer zum Erreichen einer individuell gut kontrollierten Woche 16 Tage unter
Salmeterol/Fluticasonpropionat-Behandlung, verglichen mit 23 Tagen nach einer Behandlung mit ICS.
Gesamtergebnisse der Studie:
|
Patienten, deren Asthma innerhalb von 12 Monaten *gut kontrolliert (Well Controlled - WC) und **vollständig kontrolliert (Totally Controlled - TC) wurde in Prozent | ||||
|
Behandlung vor der Studie |
Salmeterol/ Fluticasonpropionat |
Fluticasonpropionat | ||
|
WC |
TC |
WC |
TC | |
|
Kein ICS (Monotherapie mit SABA) |
78 % |
50 % |
70 % |
40 % |
|
Geringe Dosis ICS (< 500 pg BDP oder vergleichbare Dosis/T ag) |
75 % |
44 % |
60 % |
28 % |
|
Mittlere Dosis ICS (500-1000 pg BDP oder vergleichbare Dosis/T ag) |
62 % |
29 % |
47 % |
16 % |
|
Gepoolte Ergebnisse über die 3 Behandlungsstufen |
71 % |
41 % |
59 % |
28 % |
*Gut kontrolliertes Asthma: gelegentliche Symptome oder gelegentliche Anwendung von SABA oder vorhergesagte Lungenfunktion kleiner 80 % und kein Aufwachen in der Nacht, keine Exazerbationen und keine Nebenwirkungen, die eine Änderung der Therapie erfordern.
**Vollständige Asthmakontrolle: keine Symptome, keine Anwendung von SABA, Lungenfunktion größer als bzw. gleich 80 % der vorhergesagten Lungenfunktion, kein Aufwachen in der Nacht, keine Exazerbationen und keine Nebenwirkungen, die eine Änderung der Therapie erfordern.
Die Ergebnisse dieser Studie lassen darauf schließen, dass Salmeterol/Fluticasonpropionat 50pg/100pg, b.i.d. bei Patienten mit mittelschwerem, persistierendem Asthma, bei denen eine schnelle Asthmakontrolle notwendig ist, als initiale Erhaltungstherapie angesehen werden kann (siehe Abschnitt 4.2).
In einer randomisierten Doppelblindstudie mit Parallelgruppen, an der 318 Patienten mit persistierendem Asthma im Alter von > 18 Jahren teilnahmen, wurde die Sicherheit und Verträglichkeit von zwei Inhalationen Salmeterol/Fluticasonpropionat zweimal täglich (Doppeldosis) über einen Zeitraum von zwei Wochen bewertet. Die Studie zeigte, dass eine Verdoppelung der Inhalationen jeder Konzentration Salmeterol/Fluticasonpropionat über einen Zeitraum von 14 Tagen verglichen mit einer Inhalation zweimal täglich zu einer leicht erhöhten Inzidenz unerwünschter Ereignisse in Zusammenhang mit Beta-Agonisten (Tremor: 1 Patient [1 %] vs. 0, Palpitationen: 6 [3 %] vs. 1 [< 1 %], Muskelkrämpfe: 6 [3 %] vs. 1 [< 1 %]) und einer ähnlichen Inzidenz unerwünschter Ereignisse in Zusammenhang mit inhalativen Corticosteroiden (z.B. orale Candidose: 6 [6 %] vs. 16 [8 %], Heiserkeit: 2 [2 %] vs. 4 [2 %]) führte.
Die leicht erhöhte Inzidenz unerwünschter Ereignisse in Zusammenhang mit BetaAgonisten sollte berücksichtigt werden, wenn der Arzt eine Verdoppelung der Dosis Salmeterol/Fluticasonpropionat bei erwachsenen Patienten, bei denen eine zusätzliche kurzzeitige Therapie (bis zu 14 Tagen) mit inhalativen Corticosteroiden erforderlich ist, in Betracht zieht.
Klinische Studien mit Salmeterol/Fluticasonpropionat bei Patienten mit COPD
Bei TORCH handelte es sich um eine 3-jährige Studie zur Beurteilung der Wirkung einer Behandlung mit Salmeterol/Fluticasonpropionat 50 pg/500 pg, b.i.d., Salmeterol 50 pg, b.i.d., Fluticasonpropionat (FP) 500 pg, b.i.d. oder Placebo auf die Gesamtmortalität bei Patienten mit COPD. Patienten mit COPD mit einer FEV1 < 60 % des vorhergesagten Normwerts zu Studienbeginn (vor Anwendung eines
Bronchodilatators) wurden auf eine doppelt verblindete Therapie randomisiert. Während der Studie konnten die Patienten ihre gewöhnliche Therapie mit Ausnahme anderer inhalativer Corticosteroide, langwirkender Bronchodilatatoren und
systemischer Langzeit-Corticosteroide fortsetzen. Für alle Patienten wurde der Überlebensstatus nach 3 Jahren bestimmt, ungeachtet dessen, ob die Behandlung mit dem Prüfpräparat abgebrochen wurde. Als primärer Endpunkt wurde die Reduzierung der Gesamtmortalität nach 3 Jahren bei einer Behandlung mit Salmeterol/Fluticasonpropionat verglichen mit Placebo festgelegt.
|
Placebo N=1.524 |
Salmeterol 50 N=1.521 |
FP 500 N=1.534 |
Salmeterol/FP 50/500 N=1.533 | |
|
Gesamtmortalität nach 3 Jahren | ||||
|
Anzahl der Todesfälle (%) |
231 (15,2 %) |
205 (13,5 %) |
246 (16,0 %) |
193 (12,6 %) |
|
Hazard-Rate vs. Placebo (CI) |
entfällt |
0,879 (0,73; 1,06) |
1,060 (0,89; 1,27) |
0,825 (0,68; 1,00) |
|
P-Wert |
0,180 |
0,525 |
0,0521 | |
|
Hazard-Rate Salmeterol/FP 50/500 vs. Bestandteile (CI) |
entfällt |
0,932 (0,77; 1,13) |
0,774 (0,64; 0,93) |
Entfällt |
|
P-Wert |
0,481 |
0,007 | ||
|
1Nicht signifikanter P-Wert nach Anpassung für 2 Zwischenanalysen zum Vergleich der primären Wirksamkeit gemäß einer anhand des Raucherstatus stratifizierten Logrank-Analyse | ||||
Verglichen mit Placebo lag bei Patienten, die mit Salmeterol/Fluticasonpropionat über einen Zeitraum von 3 Jahren behandelt wurden, tendenziell eine verbesserte Überlebenszeit vor. Die statistische Signifikanz von p < 0,05 konnte jedoch nicht erreicht werden.
Der prozentuale Anteil von Patienten, die innerhalb von 3 Jahren aufgrund einer COPD-bedingten Ursache gestorben sind, betrug 6,0% unter Placebo, 6,1% unter Salmeterol, 6,9% unter FP und 4,7% unter Salmeterol/Fluticasonpropionat.
Der durchschnittliche Anteil mittelschwerer bis schwerer Exazerbationen pro Jahr war in der Salmeterol/Fluticasonpropionat-Gruppe verglichen mit einer Therapie mit Salmeterol, FP und Placebo signifikant reduziert (mittlere Häufigkeit in der Salmeterol/Fluticasonpropionat-Gruppe 0,85 verglichen mit 0,97 in der Salmeterol-, 0,93 in der FP- und 1,13 in der Placebo-Gruppe). Dies entspricht einer Reduzierung der Rate mittelschwerer bis schwerer Exazerbationen von 25 % (95 % CI: 19 % bis 31%; p < 0,001) verglichen mit Placebo, 12 % verglichen mit Salmeterol (95 % CI: 5 % bis 19 %; p = 0,002) und 9 % verglichen mit FP (95 % CI: 1 % bis 16 %; p < 0,024). Im Vergleich zu Placebo reduzierten Salmeterol und FP signifikant die Exazerbationsrate um 15 % (95 % CI: 7 % bis 22 %; p < 0,001) bzw. 18 % (95 % CI: 11 % bis 24 %;
p < 0,001).
Die gesundheitsbezogene Lebensqualität gemäß dem SGRQ-Fragebogen zu Atemwegsbeschwerden (St George's Respiratory Questionnaire) wurde im Vergleich zu Placebo in allen Behandlungen mit dem Verum verbessert. Die durchschnittliche Verbesserung innerhalb von 3 Jahren betrug bei Salmeterol/Fluticasonpropionat verglichen mit Placebo -3,1 Einheiten (95 % CI: -4,1 bis -2,1; p < 0,001), verglichen mit Salmeterol -2,2 Einheiten (p < 0,001) und verglichen mit FP 1,2 Einheiten (p = 0,017). Eine Verringerung um 4 Einheiten gilt als klinisch relevant.
Die geschätzte 3-Jahres-Wahrscheinlichkeit einer als unerwünschtes Ereignis berichteten Pneumonie lag bei 12,3 % für Placebo, 13,3 % für Salmeterol, 18,3 % für FP und 19,6 % für Salmeterol/Fluticasonpropionat (Hazard-Rate von Salmeterol/Fluticasonpropionat vs. Placebo: 1,64, 95 % CI: 1,33 bis 2,01, p < 0,001). Es gab keine erhöhte Mortalität aufgrund einer Pneumonie. Während der Behandlung betrug die Anzahl von hauptsächlich von einer Pneumonie verursachten Todesfälle 7 in der Placebo-, 9 in der Salmeterol-, 13 in der FP- und 8 in der Salmeterol/Fluticasonpropionat-Gruppe. Es gab keine signifikante Differenz in der Wahrscheinlichkeit einer Knochenfraktur (5,1 % Placebo, 5,1 % Salmeterol, 5,4 % FP und 6,3 % Salmeterol/ Fluticasonpropionat; Hazard-Rate für Salmeterol/Fluticasonpropionat vs. Placebo: 1,22, 95 % CI: 0,87 bis 1,72, p = 0,248).
Placebokontrollierte klinische Prüfungen über einen Zeitraum von 6 und 12 Monaten zeigten, dass die regelmäßige Anwendung von Salmeterol/Fluticasonpropionat 50 pg/500 pg die Lungenfunktion verbessert und Atemlosigkeit sowie Anwendung von Notfallmedikamenten verringert.
Die SMART-Studie (Salmeterol Multicenter Asthma Research Trial)
Bei der SMART-Studie handelte es sich um eine 28-wöchige multizentrische, randomisierte, placebokontrollierte Doppelblindstudie mit Parallelgruppen, die in den USA durchgeführt wurde und in der 13.176 Patienten auf Salmeterol (50 pg, zweimal täglich) und 13.179 Patienten auf Placebo zusätzlich zu ihrer regulären Asthmatherapie randomisiert wurden. In die Studie wurden Patienten (ab 12 Jahre), die an Asthma litten und zum Zeitpunkt der Studie ein Asthmapräparat (außer LABA) einnahmen, aufgenommen. Die Anwendung von ICS zu Studienbeginn wurde bei Aufnahme in die Studie notiert, war für die Studie jedoch nicht relevant. Als primärer Endpunkt der SMART-Studie wurde eine kombinierte Anzahl von atemwegsbedingten Todesfällen und atemwegsbedingten, lebensbedrohlichen Erfahrungen festgelegt.
Hauptergebnisse der SMART-Studie: primärer Endpunkt
|
Patientengruppe |
Anzahl der Ereignisse des primären Endpunkts/Anzahl der Patienten |
Relatives Risiko | |
|
Salmeterol |
Placebo |
(95 % Konfidenzintervall) | |
|
Alle Patienten |
50/13.176 |
36/13.179 |
1,40 (0,91; 2,14) |
|
Patienten, die inhalative Steroide anwenden |
23/6.127 |
19/6.138 |
1,21 (0,66; 2,23) |
|
Patienten, die keine inhalativen Steroide anwenden |
27/7.049 |
17/7.041 |
1,60 (0,87; 2,93) |
|
Patienten afroamerikanischer Herkunft |
20/2.366 |
5/2.319 |
4,10 (1,54; 10,90) |
(Die Risikowerte in Fettdruck haben eine statistische Signifikanz von 95 %.)
Hauptergebnisse der SMART-Studie bei Anwendung inhalativer Steroide zu Studienbeginn: sekundäre Endpunkte
|
Anzahl der Ereignisse des sekundären Endpunkts/Anzahl der Patienten |
Relatives Risiko (95 % Konfidenzintervall) | ||
|
Salmeterol |
Placebo | ||
|
Atemwegsbedingte T |
fodesfälle | ||
|
Patienten, die inhalative Steroide anwenden |
10/6.127 |
5/6.138 |
2,01 (0,69; 5,86) |
|
Patienten, die keine inhalativen Steroide anwenden |
14/7.049 |
6/7.041 |
2,28 (0,88; 5,94) |
|
Kombinierte Todesfälle oder lebensbedrohliche Erfahrungen au |
Fgrund von Asthma | ||
|
Patienten, die inhalative Steroide anwenden |
16/6.127 |
13/6.138 |
1,24, (0,60; 2,58) |
|
Patienten, die keine inhalativen Steroide anwenden |
21/7.049 |
9/7.041 |
2,39 (1,10; 5,22) |
|
Asthmabedingte Toc |
esfälle | ||
|
Patienten, die inhalative Steroide anwenden |
4/6.127 |
3/6.138 |
1,35 (0,30; 6,04) |
|
Patienten, die keine inhalativen Steroide anwenden |
9/7.049 |
0/7.041 | |
(*=konnte aufgrund fehlender Ereignisse in der Placebo-Gruppe nicht berechnet werden. Risikowerte in Fettdruck haben eine statistische Signifikanz von 95 %. Die sekundären Endpunkte aus oben genannter Tabelle erreichten in der gesamten Population eine statistische Signifikanz.) Die sekundären Endpunkte der kombinierten Gesamtzahl an Todesfällen oder lebensbedrohlichen Erfahrungen, Todesfälle bzw. Aufnahmen in ein Krankenhaus unabhängig von der Ursache erreichten in der gesamten Population keine statistische Signifikanz.
Wirkmechanismus:
Rolenium® enthält Salmeterol und Fluticasonpropionat, die jeweils einen anderen Wirkmechanismus haben. Im Folgenden sind die jeweiligen Wirkmechanismen der beiden Wirkstoffe aufgeführt:
Salmeterol:
Salmeterol ist ein selektiver lang wirkender (12 Stunden) Beta-2-Adrenozeptor-Agonist mit einer langen Seitenkette, die an die Exo-Seite des Rezeptors bindet.
Salmeterol bewirkt mit mindestens 12 Stunden eine länger anhaltende Bronchodilatation als herkömmliche kurzwirkende Beta-2-Agonisten in empfohlenen Dosierungen.
Fluticasonpropionat:
Fluticasonpropionat hat bei inhalativer Anwendung in den empfohlenen Dosierungen eine glucocorticoid-antiinflammatorische Wirkung in der Lunge. Hieraus resultieren reduzierte Asthmasymptome und Exazerbationen, ohne die bei einer systemischen Therapie mit Corticoiden auftretenden unerwünschten Ereignisse.
5.2 Pharmakokinetische Eigenschaften
Nach kombinierter inhalativer Anwendung von Salmeterol und Fluticasonpropionat war die Pharmakokinetik jedes einzelnen Wirkstoffs mit der einer separaten Anwendung vergleichbar. Daher kann bei der Beurteilung der Pharmakokinetik jeder einzelne Wirkstoff getrennt betrachtet werden.
Salmeterol:
Salmeterol wirkt lokal in der Lunge, weshalb die Plasmakonzentration keine Hinweise auf die therapeutische Wirkung gibt. Außerdem sind über die pharmakokinetischen Eigenschaften von Salmeterol nur begrenzte Daten verfügbar, da nach inhalativer Anwendung therapeutisch wirksamer Dosierungen nur geringe Plasmakonzentrationen (ca. 200 pg/ml oder weniger) erreicht werden, die technisch schwer zu bestimmen sind.
Fluticasonpropionat:
Die absolute Bioverfügbarkeit einer Einzeldosis inhalierten Fluticasonpropionats liegt bei gesunden Probanden etwa zwischen 5 - 11 % der angegebenen Dosis, abhängig vom verwendeten Inhalator. Bei Patienten mit Asthma oder COPD wurde
ein geringeres Ausmaß der systemischen Exposition von inhaliertem Fluticasonpropionat beobachtet.
Die systemische Resorption findet hauptsächlich über die Lungen statt und ist anfangs schnell und dann verzögert. Der Rest der inhalierten Dosis kann verschluckt werden, trägt jedoch aufgrund der geringen Wasserlöslichkeit und des präsystemischen Metabolismus nur minimal zur systemischen Exposition bei, was eine orale Bioverfügbarkeit von weniger als 1 % zur Folge hat. Die systemische Exposition steigt mit Erhöhung der inhalierten Dosis linear an.
Die Disposition von Fluticasonpropionat ist durch eine hohe Plasmaclearance (1.150 ml/min), ein großes Verteilungsvolumen im Steady-State (ca. 300 l) und eine terminale Halbwertszeit von ca. 8 Stunden charakterisiert.
Die Plasmaproteinbindung beträgt 91 %.
Fluticasonpropionat wird sehr schnell aus dem Körperkreislauf entfernt. Hauptsächlich geschieht dies durch Metabolisierung zu einem inaktiven Carbonsäuremetabolit durch das Cytochrom P450-Enzym CYP3A4. Andere nicht identifizierte Metaboliten werden auch im Stuhl gefunden.
Die renale Clearance von Fluticasonpropionat ist vernachlässigbar. Weniger als 5 % werden vorwiegend in Form von Metaboliten über den Urin ausgeschieden. Der Großteil der Dosis wird in Form von Metaboliten und als unveränderter Wirkstoff über den Stuhl ausgeschieden.
5.3 Präklinische Daten zur Sicherheit
Die einzigen Bedenken in Bezug auf die Sicherheit bei der Anwendung am Menschen, die sich aus tierexperimentellen Untersuchungen zur getrennten Verabreichung von Salmeterolxinafoat und Fluticasonpropionat ableiten ließen, waren mit einer verstärkten pharmakologischen Wirkung assoziierte Effekte.
In tierexperimentellen Untersuchungen zur Reproduktion zeigte sich, dass Glucocorticoide Fehlbildungen hervorrufen können (Gaumenspalten, Skelettfehlbildungen). Diese Ergebnisse aus tierexperimentellen Untersuchungen scheinen jedoch für den Menschen in Anbetracht des empfohlenen Dosisbereichs nicht relevant zu sein. Tierexperimentelle Untersuchungen mit Salmeterolxinafoat ergaben nur bei hoher Exposition Hinweise auf eine embryofetale Toxizität. Bei Ratten wurde nach gleichzeitiger Anwendung in Dosierungen, die bekanntlich mit Glucocorticoid-induzierten Anomalien verbunden sind, eine erhöhte Inzidenz von Transpositionen der Nabelschnurarterien sowie eine unvollständige Ossifikation des Hinterhauptbeins festgestellt.
6. Pharmazeutische Angaben
6.1 Liste der sonstigen Bestandteile
Lactose-Monohydrat.
6.2 Inkompatibilitäten
Nicht zutreffend.
2 Jahre.
6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung Nicht über 25 °C lagern.
6.5 Art und Inhalt des Behältnisses
Rolenium® enthält zwei Arzneimittel, verpackt als Einzeldosis in einem Blisterstreifen (Alu-Alu-Folie) mit 2 Näpfchen, die im Inhalator Elpenhaler® aufbewahrt werden.
Die Folie schützt das Pulver zur Inhalation vor der Wirkung der
Umgebungsbedingungen.
Jede Einzeldosis ist in einem Blisterstreifen mit 2 Näpfchen vordosiert.
Die Inhalatoren sind in einem Umkarton verpackt.
Eine Packung mit 30 Einzeldosen enthält einen Inhalator Elpenhaler® mit 30 Blisterstreifen (Klinikpackung).
Eine Packung mit 60 Einzeldosen enthält einen Inhalator Elpenhaler® mit 60 Blisterstreifen.
Eine Packung mit 180 Einzeldosen enthält drei Inhalatoren Elpenhaler® mit je 60 Blisterstreifen.
6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung
Der Arzt oder Apotheker zeigt dem Patienten, wie der Inhalator richtig angewendet wird.
ANWEISUNGEN FÜR ANWENDUNG UND HANDHABUNG DES ELPENHALER
Diese Anweisungen für den Patienten erläutern die richtige Inhalation der beiden Arzneimittel, die in einem Blisterstreifen mit 2 Näpfchen verpackt sind und im Elpenhaler® aufbewahrt werden.
Beschreibung
Der Elpenhaler® ist ein Inhalator mit dem zwei Arzneimittel in Pulverform gleichzeitig inhaliert werden. Die beiden Arzneimittel bilden zusammen ein Kombinationspräparat. Jedes Arzneimittel ist getrennt voneinander in einem der zwei Näpfchen des speziell gestalteten Blisterstreifens verpackt.
Der Blisterstreifen mit 2 Näpfchen enthält eine Dosis des Kombinationspräparats.
Der Elpenhaler® besteht aus 3 Teilen:


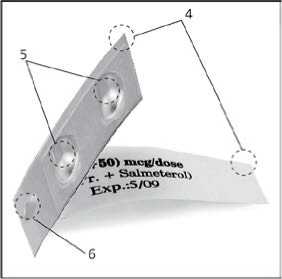
- Dem Mundstück und dessen Schutzkappe (1).
- Der Auflagefläche (2) zur Platzierung des Blisterstreifens mit 2 Näpfchen.
- Dem Aufbewahrungsfach (3) zur Unterbringung des Blisterstreifens mit 2 Näpfchen.
Die 3 Teile sind miteinander verbunden und können einzeln geöffnet werden.
Auf der Auflagefläche befindet sich:
- 1 Befestigungspunkt (2A) zur Befestigung des Blisterstreifens mit 2 Näpfchen.
- 2 Vertiefungen (2B) zur Aufnahme der zwei Näpfchen des Blisterstreifens.
- 2 Führungsstege (2C) zur sicheren Positionierung des Blisterstreifens mit 2 Näpfchen auf der Auflagefläche.
Ein Blisterstreifen mit 2 Näpfchen enthält:
- 2 Folien (4).
- 2 Näpfchen (5), in einem befindet sich Salmeterol und im anderen Fluticasonpropionat.
- 1 Loch (6).
Anwendung des Elpenhalers A. Vorbereitung des Geräts



B. Inhalation der Dosis
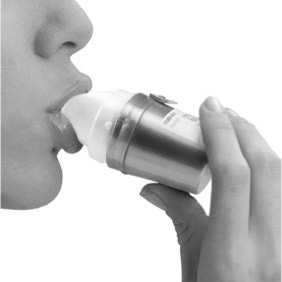
- Das Aufbewahrungsfach öffnen, einen Blisterstreifen entnehmen und das Aufbewahrungsfach wieder schließen.
Schutzkappe vollständig vom Mundstück abnehmen.
Mundstück entsperren und nach hinten klappen, um die Auflagefläche freizulegen.
Den Blisterstreifen mit 2 Näpfchen mit der glänzenden Seite nach oben halten.
Den Blisterstreifen auf den Befestigungspunkt der Auflagefläche legen. Leicht auf den Blisterstreifen drücken, um sicherzugehen, dass der Blisterstreifen am Befestigungspunkt befestigt ist.
Die 2 Näpfchen des Blisterstreifens passen nun in die dazu vorgesehenen Vertiefungen in der Auflagefläche und die Führungsstege stellen sicher, dass der Blisterstreifen richtig positioniert ist.
- Das Mundstück schließen und das überstehende Ende des Blisterstreifens abziehen. Die Dosis kann nun inhaliert werden.
Den Inhalator vom Mund entfernt halten.
- Vollständig ausatmen und darauf achten, nicht in das Mundstück des Inhalators auszuatmen.
- Den Elpenhaler® zum Mund führen und die Lippen fest um das Mundstück schließen.
- Langsam und tief durch den Mund (und nicht durch die Nase) einatmen, bis die Lunge gefüllt ist.
- Atem etwa 5 Sekunden, oder so lange, wie es angenehm ist, anhalten und gleichzeitig den Inhalator aus dem Mund nehmen.
- Ausatmen und danach normal weiteratmen.
Das Mundstück öffnen. Das gesamte Pulver wurde inhaliert und die 2 Näpfchen des Blisterstreifens sind leer.

Den leeren Blisterstreifen entfernen und mit Schritt C fortfahren.
C. Reinigen des Geräts
- Nach jedem Gebrauch das Mundstück und die Auflagefläche mit einem trockenen Tuch oder trockenen Papiertuch abwischen. Zum Reinigen des Geräts kein Wasser verwenden.
- Mundstück schließen und Schutzkappe aufsetzen.
7. Inhaber der Zulassung
ELPEN Pharmaceutical Co. Inc. 95, Marathonos Ave.
19009 Pikermi, Attica Griechenland
Mitvertrieb:
ELPEN Pharma GmbH Bismarckstr. 63 12169 Berlin Tel.: 030-797 40 40-0 Fax: 030-797 40 40-17 e-mail: info@elpen-pharma.de
8. Zulassungsnummern
Rolenium® 50 Mikrogramm/250 Mikrogramm/Dosis, einzeldosiertes Pulver zur Inhalation
81933.00. 00
Rolenium® 50 Mikrogramm/500 Mikrogramm/Dosis, einzeldosiertes Pulver zur
Inhalation
81934.00. 00
13.12.2011
10. Stand der Information
07/2013
11. Verkaufsabgrenzung
Verschreibungspflichtig
23