Spiriva Respimat 2,5 Mikrogramm Lösung Zur Inhalation
F achinformation
/^\ Boehringer n| \v Ingelheim
1. BEZEICHNUNG DES ARZNEIMITTELS
Spiriva® Respimat® 2,5 Mikrogramm Lösung zur Inhalation
2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
Die abgegebene Dosis beträgt 2,5 Mikrogramm Tiotropium pro Hub (eine therapeutische Dosis besteht aus 2 Hüben), entsprechend 3,124 Mikrogramm Tiotropiumbromid-Monohydrat.
Die abgegebene Dosis ist die Menge, die für den Patienten nach Passieren des Mundstücks verfügbar ist.
Vollständige Auflistung der sonstigen Bestandteile: siehe Abschnitt 6.1.
3. DARREICHUNGSFORM
Inhalationslösung
Klare, farblose Inhalationslösung
4. KLINISCHE ANGABEN
4.1 Anwendungsgebiete COPD
Tiotropium ist indiziert als dauerhaft einzusetzender Bronchodilatator zur Befreiung von Symptomen bei chronischer obstruktiver Lungenerkrankung (COPD).
Asthma
Spiriva Respimat ist indiziert als ein zusätzlicher dauerhaft einzusetzender Bronchodilatator bei erwachsenen Asthma-Patienten, die als Dauertherapie eine Kombination aus inhalativen Kortikosteroiden (> 800 pg Budesonid/Tag oder Äquivalent) und lang wirksamen Beta2-Agonisten erhalten, und die im Vorjahr mindestens eine schwere Exazerbation erfahren haben.
4.2 Dosierung und Art der Anwendung Dosierung
Das Arzneimittel ist nur zur Inhalation bestimmt. Die Patrone kann nur in den Spiriva Respimat Inhalator eingeführt und mit diesem verwendet werden (siehe Abschnitt 4.2 unten: Art der Anwendung).
Eine therapeutische Dosis besteht aus 2 Hüben aus dem Respimat Inhalator.
Die empfohlene Tagesdosis für Erwachsene beträgt 5 Mikrogramm Tiotropium entsprechend der Inhalation von 2 Hüben aus dem Respimat Inhalator, einmal täglich zur gleichen Tageszeit.
Die empfohlene Dosis soll nicht überschritten werden.
In der Asthma-Therapie macht sich der volle Nutzen erst nach mehreren Anwendungen des Arzneimittels bemerkbar.
Besondere Patientengruppen
Ältere Patienten können Tiotropiumbromid in der empfohlenen Dosis anwenden.
Patienten mit eingeschränkter Nierenfunktion können Tiotropiumbromid in der empfohlenen Dosis anwenden. Für Patienten mit mittlerer bis schwerer Nierenfunktionsstörung (Creatinin-Clearance < 50 ml/min) siehe Abschnitte 4.4 und 5.2.
Patienten mit eingeschränkter Leberfunktion können Tiotropiumbromid in der empfohlenen Dosis anwenden (siehe Abschnitt 5.2).
Kinder und Jugendliche
COPD
Es gibt keinen relevanten Nutzen von Spiriva Respimat bei Kindern und Jugendlichen unter 18 Jahren.
Zystische Fibrose (Mukoviszidose)
Die Sicherheit und Wirksamkeit von Spiriva Respimat ist nicht erwiesen (siehe Abschnitte 4.4 und 5.1).
Asthma
Die Sicherheit und Wirksamkeit von Spiriva Respimat ist bei Kindern und Jugendlichen noch nicht erwiesen.
Art der Anwendung
Um die richtige Anwendung des Arzneimittels zu gewährleisten, sollte dem Patienten der Gebrauch des Inhalators durch einen Arzt oder medizinisch-pharmazeutisches Fachpersonal gezeigt werden.
Gebrauchsanweisung für die Patienten
Einleitung
Lesen Sie diese Gebrauchsanweisung, bevor Sie Spiriva Respimat zum ersten Mal anwenden.
Wenden Sie diesen Inhalator einmal täglich an. Inhalieren Sie bei jeder Anwendung nacheinander 2 Hübe.
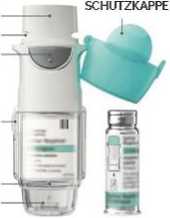
PATRONE
MUNDSTÜCK LUFTSCHLITZE AUSLÖSER
ENTRIEGELUNGSTASTE
DURCHSICHTIGES GEHÄUSEUNTERTEIL DORN ZUM AUFSTECHEN DER PATRONE
• Wenn Sie den Spiriva Respimat länger als 7 Tage nicht benutzt haben, müssen Sie zunächst einen Sprühstoß in Richtung Boden abgeben.
• Wenn Sie den Spiriva Respimat länger als 21 Tage nicht benutzt haben, müssen Sie die Schritte 4 bis 6 erneut ausführen, wie unter “Vorbereitung für die erste Anwendung” beschrieben, bis eine sichtbare Sprühwolke austritt. Wiederholen Sie dann die Schritte 4 bis 6 noch weitere 3-mal.
• Der Dom zum Aufstechen der Patrone am Boden des durchsichtigen Gehäuseunterteils darf nicht berührt werden.
Pflege des Spiriva Respimat
Reinigen Sie das Mundstück einschließlich der Metalldüse im Inneren des Mundstücks mindestens einmal wöchentlich nur mit einem feuchten Tuch oder Papiertuch.
Leichte Verfärbungen des Mundstücks haben keine Auswirkungen auf die Funktionsfähigkeit des Spiriva Respimat.
Falls erforderlich, kann die Außenseite des Spiriva Respimat Inhalators mit einem feuchten Tuch abgewischt werden.
Wann ist ein neuer Spiriva Respimat zu besorgen?
DOS1S-
A.MZEIGER

LEER
VOLL
• Bei Anwendung wie vorgesehen (2 Hübe einmal täglich) enthält der Spiriva Respimat 60 Hübe (30 Dosen).
• Der Dosisanzeiger gibt in etwa an, wie viele Hübe noch vorhanden sind.
• Wenn der Dosisanzeiger den Anfang des roten Bereiches der Skala erreicht, sind noch etwa 14 Hübe (Dosis für 7 Tage) vorhanden. Zu diesem Zeitpunkt ist die Verordnung eines neuen Spiriva Respimat notwendig.
• Sobald der Dosisanzeiger das Ende des roten Bereiches der Skala erreicht, wird der Spiriva Respimat automatisch gesperrt - es kann keine weitere Dosis abgegeben werden. Das durchsichtige Gehäuseunterteil kann nicht weiter gedreht werden.
• Der Spiriva Respimat sollte 3 Monate nach der Vorbereitung für die erste Anwendung entsorgt werden, selbst wenn er in der Zwischenzeit nicht vollständig geleert oder gar nicht angewendet wurde.
Vorbereitung für die erste Anwendung
1. Das durchsichtige Gehäuseunterteil abziehen
• Achten Sie darauf, dass die Schutzkappe geschlossen ist.
• Drücken Sie auf die Entriegelungstaste und ziehen Sie gleichzeitig mit der anderen Hand das durchsichtige Gehäuseunterteil ab.

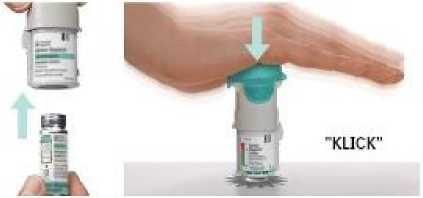
2. Die Patrone einsetzen
• Schieben Sie das schmale Ende (Oberseite) der Patrone in den Inhalator.
• Drücken Sie den Inhalator auf einer stabilen Fläche fest nach unten, bis die Patrone einrastet.
• Die Patrone darf anschließend nicht wieder entfernt werden.
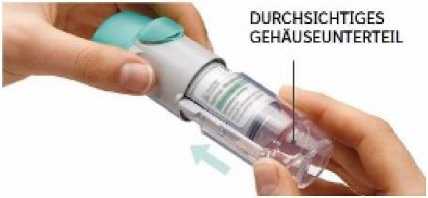
3. Das durchsichtige Gehäuseunterteil wieder aufstecken
• Stecken Sie das durchsichtige Gehäuseunterteil wieder so auf, dass es in die Entriegelungstaste einrastet.
• Das durchsichtige Gehäuseunterteil darf anschließend nicht wieder entfernt werden.
4. Drehen
• Achten Sie darauf, dass die Schutzkappe geschlossen ist.
• Drehen Sie das durchsichtige Gehäuseunterteil in Richtung der Pfeile auf dem Etikett bis es einrastet (eine halbe Umdrehung).
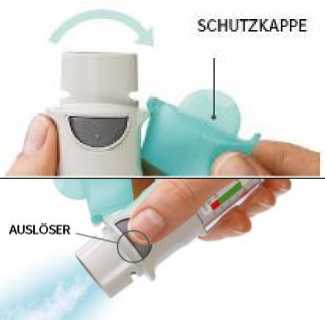
6. Auslösen
• Halten Sie den Inhalator mit der Öffnung in Richtung Boden.
• Drücken Sie den Auslöser.
• Schließen Sie die Schutzkappe.
• Wiederholen Sie die Schritte 4 bis 6 so oft, bis eine sichtbare Sprühwolke austritt.
• Wiederholen Sie dann die Schritte 4 bis 6 noch weitere 3-mal.
Der Inhalator ist nun für die Anwendung bereit. Die o. g. Schritte beeinträchtigen nicht die Anzahl der verfügbaren Dosen. Nach dieser Vorbereitung wird der Inhalator die vorgesehenen 60 Hübe (30 Dosen) abgeben._
Tägliche Anwendung

Drehen
• Achten Sie darauf, dass die Schutzkappe geschlossen ist.
• Drehen Sie das durchsichtige Gehäuseunterteil in Richtung der Pfeile auf dem Etikett bis es einrastet (eine halbe Umdrehung).
Auslösen
• Atmen Sie langsam und vollständig aus.
• Umschließen Sie das Mundstück mit den Lippen, ohne die Luftschlitze zu verdecken. Halten Sie den Inhalator waagerecht in Richtung Rachen.
• Atmen Sie langsam und tief durch den Mund ein, drücken Sie gleichzeitig den Auslöser und atmen Sie weiter langsam ein, solange es nicht unangenehm wird.
• Halten Sie den Atem möglichst
10 Sekunden lang an oder solange, dass es nicht unangenehm wird.
• Wiederholen Sie die Schritte “Drehen” -“Öffnen” - “Auslösen” einmal, um den 2. Hub zu inhalieren.
• Schließen Sie die Schutzkappe bis zum nächsten Gebrauch des Inhalators.

4.3 Gegenanzeigen
Spiriva Respimat ist kontraindiziert bei Patienten mit Überempfindlichkeit gegenüber Tiotropiumbromid, Atropin oder einem seiner Derivate, wie z. B. Ipratropium oder Oxitropium, oder gegenüber einem der sonstigen Bestandteile (siehe Abschnitt 6.1.).
4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Als Bronchodilatator zur Dauerbehandlung mit einmal täglicher Anwendung sollte Tiotropiumbromid weder zur Erstbehandlung akuter Bronchospasmen noch zur Linderung akuter Symptome eingesetzt werden. Bei einem akuten Anfall sollte ein schnell wirkender Beta2-Agonist verwendet werden.
Spiriva Respimat sollte bei Asthma nicht als Erste Wahl- bzw. Monotherapie angewendet werden. Asthmapatienten sind anzuweisen, ihre bisherige antiinflammatorische Behandlung (inhalative Kortikosteroide) nach Beginn der Therapie mit Spiriva Respimat unverändert fortzusetzen, auch bei Besserung der Symptome.
Nach der Anwendung von Tiotropiumbromid Inhalationslösung sind Immunreaktionen vom Soforttyp möglich.
Aufgrund seiner anticholinergen Aktivität sollte Tiotropiumbromid bei Patienten mit Engwinkelglaukom, Prostatahyperplasie oder Harnblasenhalsverengung nur mit Vorsicht angewendet werden.
Inhalative Arzneimittel können zu inhalationsbedingten Bronchospasmen führen.
Tiotropium sollte mit Vorsicht angewendet werden bei Patienten mit einem Myokardinfarkt innerhalb der letzten 6 Monate; mit instabilen oder lebensbedrohlichen Herzrhythmusstörungen oder Herzrhythmusstörungen, die eine Intervention oder eine Umstellung der medikamentösen Therapie erforderten, innerhalb der letzten 12 Monate; Hospitalisierung wegen Herzinsuffizienz (NYHA Grad III oder IV) innerhalb der letzten 12 Monate. Solche Patienten waren von den klinischen Prüfungen ausgeschlossen, und die genannten Erkrankungen können von der anticholinergen Wirkungsweise betroffen sein.
Da die Plasmakonzentration mit nachlassender Nierenfunktion bei Patienten mit mittlerer bis schwerer Nierenfunktionsstörung (Creatinin-Clearance < 50 ml/min) ansteigt, sollte Tiotropiumbromid nur dann angewendet werden, wenn der zu erwartende Nutzen ein potenzielles Risiko überwiegt. Langzeiterfahrungen bei Patienten mit schwerer Nierenfunktionsstörung liegen nicht vor (siehe Abschnitt 5.2).
Patienten sollten angewiesen werden, darauf zu achten, dass die Inhalationslösung nicht in die Augen gelangt. Sie sind darüber zu informieren, dass dies zum Auftreten oder zur Verschlimmerung eines Engwinkelglaukoms, Augenschmerzen oder Missempfinden, vorübergehend verschwommenem Sehen, Augenhalos oder unwirklichem Farbempfinden in Verbindung mit geröteten Augen durch Blutstauungen in der Bindehaut und Hornhautödem führen kann. Wenn zwei oder mehrere dieser Symptome gleichzeitig auftreten, sollte die Anwendung von Tiotropiumbromid abgebrochen und unverzüglich ein Augenarzt aufgesucht werden.
Mundtrockenheit, wie sie unter Therapie mit Anticholinergika beobachtet wurde, könnte bei längerer Dauer zum Auftreten von Karies führen.
Tiotropiumbromid sollte nicht häufiger als einmal täglich angewendet werden (siehe Abschnitt 4.9).
Bei zystischer Fibrose (Mukoviszidose) wird die Anwendung von Spiriva Respimat nicht empfohlen. Wenn Spiriva Respimat bei Patienten mit zystischer Fibrose angewendet wird, kann dies die Symptome der zystischen Fibrose verstärken (z. B. schwerwiegende unerwünschte Ereignisse, pulmonale Exazerbationen, Atemwegsinfekte).
4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Obwohl keine gezielten Untersuchungen zu Wechselwirkungen mit Arzneimitteln durchgeführt wurden, wurde Tiotropiumbromid zusammen mit anderen Arzneimitteln ohne Anzeichen von Arzneimittel-Wechselwirkungen angewendet. Bei diesen Arzneimitteln handelt es sich u. a. um sympathomimetische Bronchodilatatoren, Methylxanthine, orale und inhalative Steroide, Antihistaminika, Mukolytika, Leukotrien-Moderatoren, Mastzellstabilisatoren (Cromone) und Anti-IgE-Therapie, die üblicherweise bei der Behandlung von COPD und Asthma angewendet werden.
Die Anwendung von LABA oder ICS ändert die Exposition gegenüber Tiotropium nicht.
Die gleichzeitige Anwendung von Tiotropiumbromid und anderen Anticholinergika wurde nicht untersucht und wird daher nicht empfohlen.
4.6 Fertilität, Schwangerschaft und Stillzeit
Schwangerschaft
Bisher liegen nur sehr begrenzte Erfahrungen in der Anwendung von Tiotropium bei Schwangeren vor. Tierexperimentelle Studien ergaben keine Hinweise auf direkte oder indirekte gesundheitsschädliche Wirkungen in Bezug auf eine Reproduktionstoxizität in klinisch relevanten Dosen (siehe Abschnitt 5.3). Vorsichtshalber soll eine Anwendung von Spiriva Respimat während der Schwangerschaft vermieden werden.
Stillzeit
Es ist nicht bekannt, ob Tiotropiumbromid beim Menschen in die Muttermilch ausgeschieden wird. Obwohl Studien mit Nagetieren gezeigt haben, dass Tiotropiumbromid nur in geringer Menge in die Muttermilch ausgeschieden wird, sollte Spiriva Respimat während der Stillzeit nicht angewendet werden. Tiotropiumbromid ist eine langwirksame Substanz. Die Entscheidung, ob das Stillen oder die Behandlung mit Spiriva Respimat beendet oder beides fortgeführt werden soll, sollte unter Berücksichtigung der Vorteile des Stillens für das Kind und der Behandlung mit Spiriva Respimat für die Stillende getroffen werden.
Fertilität
Klinische Daten zur Fertilität liegen für Tiotropium nicht vor. Eine nicht-klinische Studie mit Tiotropium zeigte keinen Hinweis auf negative Auswirkungen auf die Fertilität (siehe Abschnitt 5.3).
4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Es wurden keine Untersuchungen über die Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen durchgeführt. Beim Auftreten von Schwindel oder verschwommenem Sehen kann die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen beeinträchtigt sein.
4.8 Nebenwirkungen
Zusammenfassung des Sicherheitsprofils
Viele der aufgeführten Nebenwirkungen können auf die anticholinergen Eigenschaften von Tiotropiumbromid zurückgeführt werden.
Tabellarische Zusammenfassung der Nebenwirkungen
Die Häufigkeiten der unten aufgelisteten Nebenwirkungen basieren auf dem Vorkommen dieser unerwünschten Arzneimittelwirkungen (Ereignisse, die Tiotropium zugeordnet werden), die in den gepoolten Tiotropiumgruppen von 7 Placebo-kontrollierten klinischen Studien bei COPD (3.282 Patienten) und 6 Placebo-kontrollierten klinischen Studien bei Asthma (1.256 Patienten), mit einer Behandlungsdauer von 4 Wochen bis zu einem Jahr, beobachtet wurden.
Bei den Häufigkeitsangaben zu Nebenwirkungen werden folgende Kategorien zugrunde gelegt:
Sehr häufig (> 1/10); Häufig (> 1/100 bis < 1/10); Gelegentlich (> 1/1.000 bis < 1/100); Selten (> 1/10.000 bis < 1/1.000); Sehr selten (< 1/10.000); Nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar)
|
Systemorganklasse / MedDRA-Terminologie |
Häufigkeit bei COPD |
Häufigkeit bei Asthma |
|
Stoffwechsel- und Ernährungsstörungen | ||
|
Dehydrierung |
nicht bekannt |
nicht bekannt |
|
Erkrankungen des Nervensystems | ||
|
Schwindel |
gelegentlich |
gelegentlich |
|
Kopfschmerzen |
gelegentlich |
gelegentlich |
|
Insomnie |
selten |
gelegentlich |
|
Augenerkrankungen | ||
|
Glaukom |
selten |
nicht bekannt |
|
Erhöhter Augeninnendruck |
selten |
nicht bekannt |
|
Verschwommenes Sehen |
selten |
nicht bekannt |
|
Herzerkrankungen | ||
|
V orhofflimmern |
selten |
nicht bekannt |
|
Palpitationen |
selten |
gelegentlich |
|
Supraventrikuläre Tachykardien |
selten |
nicht bekannt |
|
Tachykardie |
selten |
nicht bekannt |
|
Erkrankungen der Atemwege, des Brustraums und des Mediastinums | ||
|
Husten |
gelegentlich |
gelegentlich |
|
Pharyngitis |
gelegentlich |
gelegentlich |
|
Dysphonie |
gelegentlich |
gelegentlich |
|
Epistaxis |
selten |
nicht bekannt |
|
Bronchospasmus |
selten |
gelegentlich |
|
Laryngitis |
selten |
nicht bekannt |
|
Sinusitis |
nicht bekannt |
nicht bekannt |
|
Erkrankungen des Gastrointestinaltrakts | ||
|
Mundtrockenheit |
häufig |
häufig |
|
Obstipation |
gelegentlich |
selten |
|
Oropharyngeale Candidose |
gelegentlich |
gelegentlich |
|
Dysphagie |
selten |
nicht bekannt |
|
Gastro-ösophagealer Reflux |
selten |
nicht bekannt |
|
Karies |
selten |
nicht bekannt |
|
Gingivitis |
selten |
selten |
|
Glossitis |
selten |
nicht bekannt |
|
Stomatitis |
nicht bekannt |
selten |
|
Systemorganklasse / MedDRA-Terminologie |
Häufigkeit bei COPD |
Häufigkeit bei Asthma |
|
Intestinale Obstruktion, einschließlich paralytischem Ileus |
nicht bekannt |
nicht bekannt |
|
Übelkeit |
nicht bekannt |
nicht bekannt |
|
Erkrankungen der Haut und des Unterhautzellgewebes, Störungen des Immunsystems | ||
|
Hautausschlag (Rash) |
gelegentlich |
selten |
|
Pruritus |
gelegentlich |
selten |
|
Angioneurotisches Ödem |
selten |
selten |
|
Urtikaria |
selten |
selten |
|
Hautinfektionen, Hautulkus |
selten |
nicht bekannt |
|
Trockene Haut |
selten |
nicht bekannt |
|
Überempfindlichkeitsreaktionen (einschließlich Reaktionen vom Soforttyp) |
nicht bekannt |
selten |
|
Anaphylaktische Reaktion |
nicht bekannt |
nicht bekannt |
|
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen | ||
|
Gelenkschwellung |
nicht bekannt |
nicht bekannt |
|
Erkrankungen der Nieren und Harnwege | ||
|
Harnverhalt |
gelegentlich |
nicht bekannt |
|
Dysurie |
gelegentlich |
nicht bekannt |
|
Harnwegsinfekte |
selten |
nicht bekannt |
Angaben zu ausgewählten Nebenwirkungen
In kontrollierten klinischen Prüfungen mit COPD-Patienten wurden unter den Nebenwirkungen am häufigsten anticholinerge Effekte beobachtet, wie z. B. Mundtrockenheit (bei ca. 2,9 % der Patienten). Bei Asthma-Patienten betrug die Inzidenz für Mundtrockenheit 1,2 %.
In 7 klinischen Prüfungen mit COPD-Patienten brachen 3 von 3.282 mit Tiotropium behandelten Patienten (0,1 %) die Studie wegen Mundtrockenheit ab. In 6 klinischen Prüfungen mit 1.256 AsthmaPatienten wurden keine Studienabbrüche aufgrund von Mundtrockenheit gemeldet.
Zu schwerwiegenden Nebenwirkungen aufgrund des anticholinergen Effektes gehören Glaukom, Verstopfung und Darmobstruktion einschließlich paralytischem Ileus, sowie Harnverhalt.
Besondere Patientengruppen
Mit fortgeschrittenem Alter ist eine Zunahme der anticholinergen Effekte möglich.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Arzneimittel und Medizinprodukte,
Abt. Pharmakovigilanz, Kurt-Georg-Kiesinger-Allee 3, D-53175 Bonn,
Website: www.bfarm.de anzuzeigen.
4.9 Überdosierung
In hohen Dosen kann Tiotropiumbromid zu anticholinergen Symptomen führen.
Bei gesunden Probanden traten jedoch keine systemischen anticholinergen Nebenwirkungen nach der Inhalation einer Einzeldosis von bis zu 340 Mikrogramm Tiotropiumbromid auf. Bei einer Dosierung von bis zu 40 Mikrogramm Tiotropium Inhalationslösung über 14 Tage wurden bei den gesunden
Probanden außer Mund- und Rachentrockenheit sowie trockener Nasenschleimhaut (deutliche Reduzierung des Speichelflusses ab dem 7. Tag) keine relevanten Nebenwirkungen beobachtet.
5. PHARMAKOLOGISCHE EIGENSCHAFTEN
5.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Andere inhalative Mittel bei obstruktiven Atemwegserkrankungen,
Anticholinergika
ATC-Code: R03BB04
Wirkmechanismus
Tiotropium ist ein langwirksamer, spezifischer Muskarinrezeptor-Antagonist. Es weist eine ähnliche Affinität zu den Muskarinrezeptor-Subtypen M1 bis M5 auf. In den Atemwegen bindet Tiotropiumbromid kompetitiv und antagonistisch reversibel an den M3-Rezeptoren der glatten Bronchialmuskulatur; antagonisiert dort die cholinerge (bronchokonstriktive) Wirkung von Acetylcholin, was zu einer Relaxation der glatten Bronchialmuskulatur führt. Die Wirkung war dosisabhängig und hielt länger als 24 Stunden an. Als N-quartäres Anticholinergikum ist Tiotropiumbromid nach inhalativer Applikation topisch (broncho-) selektiv und zeigt eine akzeptable therapeutische Breite, ehe es zu systemischen anticholinergen Wirkungen kommt.
Pharmakodynamische Wirkungen
Tiotropium dissoziiert insbesondere von M3-Rezeptoren sehr langsam und weist eine signifikant längere Dissoziationshalbwertszeit als Ipratropium auf. Die Dissoziation von M2-Rezeptoren ist schneller als die von M3-Rezeptoren, was sich in in-vitro-Studien funktionell als kinetisch kontrollierte Rezeptorsubtypenselektivität von M3 gegenüber M2 zeigte. Die hohe Wirkstärke, sehr langsame Rezeptordissoziation und Bronchoselektivität aufgrund topischer, inhalativer Anwendung korreliert klinisch mit signifikanter und lang wirkender Bronchodilatation bei Patienten mit COPD und Asthma.
Klinische Wirksamkeit und Sicherheit bei COPD
Das klinische Phase-III-Entwicklungsprogramm umfasste zwei 1-jährige, zwei 12-wöchige und zwei 4-wöchige randomisierte, doppelblinde Studien an 2.901 COPD-Patienten (von denen 1.038 eine Dosis von 5 Mikrogramm Tiotropium erhielten). Die 1-Jahres-Programme bestanden aus zwei Placebo-kontrollierten Prüfungen. Die beiden 12-Wochen-Studien waren sowohl aktiv (Ipratropium-) als auch Placebo-kontrolliert. In allen sechs Studien wurde die Lungenfunktion untersucht. Zusätzlich wurden in den beiden 1-jährigen Studien die Entwicklung von Dyspnoe, gesundheitsbezogener Lebensqualität und der Einfluss auf Exazerbationen untersucht.
Placebo-kontrollierte Studien
Lungenfunktion
Die einmal tägliche Anwendung von Tiotropium Inhalationslösung führte innerhalb von 30 Minuten nach der ersten Dosis zu einer signifikanten Verbesserung der Lungenfunktion (forcierte exspiratorische Einsekundenkapazität (FEV1) und forcierte Vitalkapazität (FVC) im Vergleich zu Placebo (durchschnittliche FEV1-Verbesserung nach 30 Minuten: 0,113 l; 95 % Konfidenzintervall (KI): 0,102-0,125 l; p < 0,0001). Diese hielt im Steady State für die Dauer von 24 Stunden an: durchschnittliche FEVi-Verbesserung im Vergleich zu Placebo 0,122 l; 95 % KI: 0,106-0,138 l;
p < 0,0001.
Der pharmakodynamische Steady State wurde innerhalb von einer Woche erreicht.
Spiriva Respimat verbesserte signifikant den morgendlichen und abendlichen PEFR (Peak Flow Wert) gemäß den täglichen Aufzeichnungen der Patienten im Vergleich zu Placebo (durchschnittliche PEFR-Verbesserung: durchschnittliche Verbesserung am Morgen 22 l/min; 95 % KI: 18-55 l/min,
p < 0,0001; abends 26 l/min; 95 % KI: 23-30 l/min, p < 0,0001). Die Anwendung von Spiriva Respimat führte im Vergleich zu Placebo zu einer Reduktion an bronchodilatatorischer Notfallmedikation (durchschnittliche Verbesserung in der Anwendung nach Bedarf um 0,66 Anwendungen pro Tag, 95 % KI: 0,51-0,81 Anwendungen pro Tag, p < 0,0001).
Die bronchodilatatorische Wirkung von Spiriva Respimat blieb ohne Anzeichen einer Toleranzentwicklung über ein Jahr Anwendungsdauer bestehen.
Dyspnoe, gesundheitsbezogene Lebensqualität, COPD-Exazerbationen in Langzeitstudien über 1 Jahr Dyspnoe
Spiriva Respimat verbesserte signifikant die Dyspnoe (ausgewertet mittels Transition Dyspnoea Index) im Vergleich zu Placebo (durchschnittliche Verbesserung 1,05 Einheiten; 95 % KI:
0,73-1,38 Einheiten, p < 0,0001). Die Verbesserung blieb während des gesamten Behandlungszeitraumes bestehen.
Gesundheitsbezogene Lebensqualität
Die vom Patienten mit dem St. George’s Respiratory Questionnaire bewertete Lebensqualität verbesserte sich mit Spiriva Respimat im Vergleich zu Placebo am Ende der zwei 1-Jahres-Studien um 3,5 Einheiten (95 % KI: 2,1-4,9 Einheiten, p < 0,0001). Eine Verminderung um 4 Einheiten wird als klinisch relevante Verbesserung angesehen.
COPD-Exazerbationen
In drei randomisierten, doppelblinden, Placebo-kontrollierten 1-Jahres-Studien führte die Behandlung mit Spiriva Respimat zu einem signifikant reduzierten Risiko einer COPD-Exazerbation im Vergleich zu Placebo. COPD-Exazerbationen wurden definiert als „ein Komplex bestehend aus mindestens zwei respiratorischen Ereignissen/Symptomen über eine Dauer von drei Tagen oder mehr, mit der Notwendigkeit einer Änderung der Behandlung (Verschreibung von Antibiotika und/oder systemischen Kortikosteroiden und/oder eine bedeutende Änderung der verschriebenen Atemwegsmedikamente)“.
Die Behandlung mit Spiriva Respimat führte zu einem reduzierten Risiko einer Hospitalisierung aufgrund einer COPD-Exazerbation (signifikant in einer entsprechend gepowerten großen Exazerbationsstudie).
Die gepoolte Analyse von zwei Phase-III-Studien und die separate Analyse einer zusätzlichen Exazerbationsstudie werden in Tabelle 1 gezeigt. Alle Atemwegsmedikamente außer Anticholinergika und langwirksamen Beta-Agonisten waren als Begleitmedikation erlaubt, d. h. schnell wirksame Beta-Agonisten, inhalative Kortikosteroide und Xanthine. Langwirksame Beta-Agonisten waren in der Exazerbationsstudie zusätzlich erlaubt.
Tabelle 1: Statistische Analyse der COPD-Exazerbationen und der Hospitalisierungen aufgrund von COPD-Exazerbationen bei Patienten mit mittelgradiger bis sehr schwerer COPD
|
Studie (N^^iva:; NPlacebo) |
Endpunkt |
Spiriva Respimat |
Placebo |
% Risikoreduktion (95 % KI)a |
p-Wert |
|
1-jährige Phase-III-Studien, gepoolte Analyse d (670,653) |
Tage bis zur ersten COPD-Exazerbation |
160 a |
86 a |
29 (16 bis 40)b |
< 0,0001 b |
|
Mittlere Inzidenzrate der Exazerbationen pro Patientenjahr |
0,78 c |
1,00 c |
22 (8 bis 33)c |
0,002 c | |
|
Zeit bis zur ersten Hospitalisierung aufgrund einer COPD-Exazerbation |
25 (-16 bis 51)b |
0,20 b |
|
Mittlere Inzidenzrate der Hospitalisierungen aufgrund von Exazerbationen pro Patientenjahr |
0,09 c |
0,11 c |
20 (-4 bis 38)c |
0,096 c | |
|
1-jährige Phase-IIIb- Exazerbations studie (1.939, 1.953) |
Tage bis zur ersten COPD-Exazerbation |
169 a |
119 a |
31 (23 bis 37) b |
< 0,0001 b |
|
Mittlere Inzidenzrate der Exazerbationen pro Patientenjahr |
0,69 c |
0,87 c |
21 (13 bis 28) c |
< 0,0001 c | |
|
Zeit bis zur ersten Hospitalisierung aufgrund einer COPD-Exazerbation |
27 (10 bis 41)b |
0,003 b | |||
|
Mittlere Inzidenzrate der Hospitalisierungen aufgrund von Exazerbationen pro Patientenjahr |
0,12 c |
0,15 c |
19 (7 bis 30)c |
0,004 c |
a Zeit bis zum ersten Ereignis: Behandlungstage, nach denen bei 25 % der Patienten mindestens eine COPD-Exazerbation/Hospitalisierung aufgrund einer Exazerbation auftrat.
In Studie A trat bei 25 % der Placebo-Patienten eine Exazerbation bis zum Tag 112 auf, während unter Spiriva Respimat bei 25 % der Patienten eine Exazerbation bis zum Tag 173 auftrat (p = 0,09).
In der Studie B trat bei 25 % der Placebo-Patienten eine Exazerbation bis zum Tag 74 auf, während unter Spiriva Respimat bei 25 % der Patienten eine Exazerbation bis zum Tag 149 auftrat
(p < 0,0001).
b Hazard Ratios wurden anhand eines Cox Proportional Hazard Modell geschätzt. Der Prozentsatz der Risikoreduktion beträgt 100 x (1 - Hazard Ratio). c Poisson Regression. Risikoreduktion beträgt 100 x (1 - Rate Ratio). d Das Poolen wurde beim Studiendesign spezifiziert. Die Exazerbationsendpunkte wurden in Einzelanalysen der beiden 1-Jahres-Studien signifikant verbessert.
Aktiv-kontrollierte Tiotropium-Langzeitstudie
Zum Vergleich der Wirksamkeit und Sicherheit von Spiriva Respimat und Spiriva HandiHaler wurde eine große randomisierte, doppelblinde, aktiv-kontrollierte Langzeitstudie mit einer Beobachtungsdauer bis zu 3 Jahren durchgeführt. 5.711 Patienten erhielten Spiriva Respimat,
5.694 Patienten erhielten Spiriva HandiHaler. Die primären Endpunkte waren: Zeit bis zur ersten COPD-Exazerbation, Zeit bis zum Tod (beliebiger Ursache), sowie in einer Substudie mit 906 Patienten die FEVi-Tiefstwerte vor der nächsten Anwendung.
Die Zeit bis zur ersten COPD-Exazerbation war unter Spiriva Respimat und unter Spiriva HandiHaler numerisch vergleichbar (Hazard Ratio Spiriva Respimat/Spiriva HandiHaler: 0,98;
95 % KI: 0,93-1,03). Die mediane Anzahl der Tage bis zur ersten COPD-Exazerbation betrug bei Spiriva Respimat 756 Tage und bei Spiriva HandiHaler 719 Tage.
Die bronchodilatatorische Wirkung von Spiriva Respimat hielt über 120 Wochen an, vergleichbar zum Spiriva HandiHaler. Der mittlere Unterschied im FEV1-Tiefstwert von Spiriva Respimat versus Spiriva HandiHaler betrug -0,010 l (95 % KI: -0,038 bis +0,018 l).
In der Post-Marketing-Studie TIOSPIR zum Vergleich von Spiriva Respimat und Spiriva HandiHaler war die Gesamtmortalität, einschließlich Nachverfolgung des Vitalstatus, vergleichbar (Hazard Ratio Spiriva Respimat/Spiriva HandiHaler: 0,96; 95 % KI: 0,84-1,09), bei einer Behandlungsdauer von 13.135 bzw. 13.050 Patientenjahren.
In den Placebo-kontrollierten Studien mit Nachverfolgung des Vitalstatus bis zum Ende der vorgesehenen Behandlungsperiode ergab sich unter der Therapie mit Spiriva Respimat eine numerische Zunahme der Gesamtmortalität im Vergleich zu Placebo (Rate Ratio: 1,33;
95 % KI: 0,93-1,92), bei einer Behandlungsdauer mit Spiriva Respimat von 2.574 Patientenjahren. Die erhöhte Mortalität wurde bei Patienten mit bekannter Herzrhythmusstörung beobachtet. Unter der Therapie mit Spiriva HandiHaler ergab sich eine Reduktion des Mortalitätsrisikos um 13 % (Hazard Ratio Tiotropium/Placebo, einschließlich Nachverfolgung des Vitalstatus: 0,87; 95 % KI: 0,76-0,99), bei einer Behandlungsdauer mit Spiriva HandiHaler von 10.927 Patientenjahren. Weder in der Placebo-kontrollierten Studie mit Spiriva HandiHaler noch in der TIOSPIR-Studie zum Vergleich von Spiriva Respimat und Spiriva HandiHaler wurde in der Subgruppe der Patienten mit bekannter Herzrhythmusstörung ein erhöhtes Mortalitätsrisiko beobachtet.
Klinische Wirksamkeit und Sicherheit bei Asthma
Das klinische Phase-III-Studienprogramm zu persistierendem Asthma schloss zwei 1-Jahres-Studien, jeweils randomisiert, doppelblind und Placebo-kontrolliert, mit insgesamt 907 Asthma-Patienten (von denen 453 mit Spiriva Respimat behandelt wurden) ein, die eine Kombination aus inhalativen Kortikosterioden (ICS, > 800 pg Budesonid/Tag oder Äquivalent) und lang wirksamen Beta2-Agonisten (LABA) erhielten. Die Studien erfassten Lungenfunktions-Messwerte und schwere Exazerbationen als primäre Endpunkte.
PrimoTinA-Asthma-Studien
In den zwei 1-Jahres-Studien an Patienten, die unter einer Dauertherapie mit mindestens ICS (> 800 pg Budesonid/Tag oder Äquivalent) plus LABA symptomatisch blieben, führte die zusätzliche Therapie mit Spiriva Respimat im Vergleich zu Placebo zu klinisch relevanten Verbesserungen der Lungenfunktion.
In Woche 24 betrugen die mittleren Verbesserungen der Peak- und Trough-Werte des FEV1 0,110 l (95 % KI: 0,063-0,158 l; p < 0,0001) bzw. 0,093 l (95 % KI: 0,050-0,137 l; p < 0,0001). Die Verbesserung der Lungenfunktion im Vergleich zu Placebo hielt 24 Stunden an.
In den PrimoTinA-Asthma-Studien reduzierte die Behandlung symptomatischer Patienten mit ICS plus LABA plus Tiotropium (N=453) im Vergleich zur Behandlung symptomatischer Patienten mit ICS plus LABA plus Placebo (N=454) das Risiko schwerer Asthma-Exazerbationen um 21 %. Die Risiko-Reduktion bei der mittleren Anzahl schwerer Asthma-Exazerbationen pro Patientenjahr betrug 20 %.
Dies wurde durch eine 31 %ige Reduktion des Risikos für Asthma-Verschlechterungen sowie eine 24 %ige Risiko-Reduktion bei der mittleren Anzahl von Asthma-Verschlechterungen pro Patientenjahr gestützt (siehe Tabelle 2).
Tabelle 2: Exazerbationen bei Patienten mit Symptomen unter ICS (> 800 pg Budesonid/Tag oder Äquivalent) plus LABA (PrimoTinA-Asthma-Studien)
|
Studie |
Endpunkt |
Spiriva Respimat, zusätzlich zu mindestens ICS a plus LABA (N=453) |
Placebo, zusätzlich zu mindestens ICS a plus LABA (N=454) |
% Risikoreduktion (95 % KI) |
p-Wert |
|
zwei 1-Jahres- Studien, Phase III, gepoolte Analyse |
Tage bis zur 1. schweren Asthma-Exazerbation |
282 c |
226 c |
2F (0; 38) |
0,0343 |
|
Mittl. Anzahl schwerer Asthma-Exazerbationen / Patientenjahr |
0,530 |
0,663 |
20 d (0; 36) |
0,0458 | |
|
Tage bis zur 1. Verschlechterung des Asthmas |
315 c |
181 c |
31 b (18; 42) |
< 0,0001 | |
|
Mittl. Anzahl der AsthmaVerschlechterungen / Patientenjahr |
2,145 |
2,835 |
24^ (9; 37) |
0,0031 |
a > 800 pg Budesonid/Tag oder Äquivalent
b Hazard Ratio, Konfidenzintervall und p-Wert wurden mit einem proportionalen Hazard-Modell nach Cox geschätzt, wobei nur die Behandlung als Einfluss berücksichtigt wurde. Der Prozentsatz der Risikoreduktion beträgt 100 x (1 - Hazard Ratio)
c Zeit bis zum ersten Ereignis: Behandlungstage, nach denen bei 25 % / 50 % der Patienten mindestens eine schwere Asthma-Exazerbation bzw. Verschlechterung des Asthmas auftrat
d Die Rate Ratio wurde aus einer Poisson-Regression mit LOG-Exposition (in Jahren) als Offset berechnet. Die Risikoreduktion in Prozent ist 100 x (1 - Rate Ratio)
Kinder und Jugendliche
COPD
Die Europäische Arzneimittel-Agentur hat für Spiriva Respimat eine Freistellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien bei COPD in allen pädiatrischen Altersklassen gewährt (siehe Abschnitt 4.2 bezüglich Informationen zur Anwendung bei Kindern und Jugendlichen).
Asthma
Die Europäische Arzneimittel-Agentur hat für Spiriva Respimat eine Zurückstellung der Verpflichtung zur Vorlage von Ergebnissen zu Studien bei der Asthma-Behandlung in einer oder mehreren pädiatrischen Altersklassen gewährt (siehe Abschnitt 4.2 bezüglich Informationen zur Anwendung bei Kindern und Jugendlichen).
Klinische Wirksamkeit und Sicherheit bei zystischer Fibrose
Zum klinischen Entwicklungsprogramm bei zystischer Fibrose gehörten 3 Multizenterstudien mit 959 Patienten im Alter ab 5 Monaten. Patienten unter 5 Jahren benutzten einen Spacer (AeroChamber Plus®) mit Gesichtsmaske; sie wurden nur zur Prüfung von Sicherheitsaspekten einbezogen. In den beiden Schlüsselstudien (eine Phase-II-Dosisfindungsstudie und eine konfirmatorische Phase-III-Studie) wurden die Effekte auf die Lungenfunktion (FEV1 AUC0-4h und Tiefstwert FEVi, jeweils Prozent vom Soll) von Spiriva Respimat (5 pg Tiotropium: 469 Patienten) gegen Placebo (315 Patienten) über 12 Wochen randomisiert und doppelblind verglichen. Die Phase-III-Studie umfasste auch eine offene Anschlussstudie von bis zu 12 Monaten Dauer. In diesen Studien waren alle Atemwegs-Therapeutika - mit Ausnahme von Anticholinergika - als Begleitmedikation erlaubt, z. B. langwirksame Beta-Agonisten, Mukolytika und Antibiotika.
Effekte auf die Lungenfunktion sind in Tabelle 3 dargestellt. Es wurden keine signifikanten Verbesserungen in Bezug auf Symptome und Gesundheitszustand beobachtet (Bewertung von Exazerbationen mittels „Respiratory and Systemic Symptoms Questionnaire“, Bewertung der Lebensqualität mittels „Cystic Fibrosis Questionnaire“).
Tabelle 3: Adjustierte mittlere Abweichung vom Placebo-Wert, absolute Änderungen gegenüber Baseline nach 12 Wochen
|
Phase II |
Phase III | |||||
|
Alle Patienten (NSpiriva = 176, NPlacebo 168) |
Alle Patienten (NSpiriva = 293, NPlacebo = 147) |
< 11 Jahre (NSpiriva = 95, NPlacebo = 47) |
> 12 Jahre (NSpiriva = 198, NPlacebo = 100) | |||
|
Mittelwert (95 % KI) |
p-Wert |
Mittelwert (95 % KI) |
p-Wert |
Mittelwert (95 % KI) |
Mittelwert (95 % KI) | |
|
FEV1 AUCo-4h (% vom Soll) a absolute Änderungen |
3,39 (1,67; 5,12) |
< 0,001 |
1,64 (- 0,27; 3,55) |
0,092 |
- 0,63 (- 4,58; 3,32) |
2,58 (0,50; 4,65) |
|
FEV1 AUCo-4h (Liter) absolute Änderungen |
0,09 (0,05; 0,14) |
< 0,001 |
0,07 (0,02; 0,12) |
0,010 |
0,01 (- 0,07; 0,08) |
0,10 (0,03; 0,17) |
|
Tiefstwert FEVi (% vom Soll) a absolute Änderungen |
2,22 (0,38; 4,06) |
0,018 |
1,40 (- 0,50; 3,30) |
0,150 |
- 1,24 (- 5,20; 2,71) |
2,56 (0,49; 4,62) |
|
Tiefstwert FEV1 (Liter) absolute Änderungen |
0,06 (0,01; 0,11) |
0,028 |
0,07 (0,02; 0,12) |
0,012 |
- 0,01 (- 0,08; 0,06) |
0,10 (0,03; 0,17) |
Kombinierte primäre Endpunkte
Alle Nebenwirkungen, die in den Zystische-Fibrose-Studien gemeldet wurden, sind bekannte Nebenwirkungen von Tiotropium (siehe Abschnitt 4.8). Die häufigsten Nebenwirkungen während der 12-wöchigen doppelblinden Studiendauer waren Husten (4,1 %) und Mundtrockenheit (2,8 %).
Anzahl und Prozentsatz der Patienten, die unerwünschte Ereignisse von substanzspezifischem Interesse bei zystischer Fibrose berichteten, sind in Tabelle 4 aufgeführt (ungeachtet eines kausalen Zusammenhangs). Unter der Anwendung von Tiotropium nahmen die Symptome, die als Manifestationen der zystischen Fibrose angesehen werden, numerisch, allerdings nicht statistisch signifikant, zu. Dies betraf insbesondere Patienten im Alter von < 11 Jahren.
Tabelle 4: Prozentsatz der Patienten mit unerwünschten Ereignissen von substanzspezifischem Interesse bei zystischer Fibrose während der 12-wöchigen Behandlungsphase, nach Altersgruppen, ungeachtet eines kausalen Zusammenhangs (gepoolte Werte der Phase-II- und Phase-III-Studien)
|
< 11 Jahre |
> 12 Jahre | |||
|
NPlacebo 96 |
NSpiriva — 158 |
NPlacebo 215 |
NSpiriva — 307 | |
|
Bauchschmerzen |
7,3 |
7,0 |
5,1 |
6,2 |
|
Obstipation |
1,0 |
1,9 |
2,3 |
2,6 |
|
Distales intestinales Obstruktions-Syndrom |
0 |
0 |
1,4 |
1,3 |
|
Atemwegsinfektionen |
34,4 |
36,7 |
28,4 |
28,3 |
|
Verstärkte Bronchialsekretion |
1,0 |
5,1 |
5,6 |
6,2 |
|
Exazerbationen |
10,4 |
14,6 |
18,6 |
17,9 |
„Distales intestinales Obstruktions-Syndrom” und „verstärkte Bronchialsekretion” sind „MedDRA preferred terms“. „Atemwegsinfektionen“ ist ein „MedDRA higher level group term“. „Bauchschmerzen“, „Obstipation“ und „Exazerbationen” sind Zusammenfassungen mehrerer „MedDRA preferred terms”.
Von den auf Placebo randomisierten Patienten waren 34 (10,9 %) von einem schwerwiegenden unerwünschten Ereignis betroffen, von den auf Spiriva Respimat randomisierten Patienten 56 (12,0 %).
Die Europäische Arzneimittel-Agentur hat für Spiriva Respimat eine Freistellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien bei Kindern im Alter unter 1 Jahr gewährt.
5.2 Pharmakokinetische Eigenschaften
a) Allgemeine Einleitung
Tiotropiumbromid ist eine nicht chirale quartäre Ammonium-Verbindung und in Wasser nur schwer löslich. Tiotropiumbromid wird als Inhalationslösung mittels Respimat Inhalator angewendet. Annähernd 40 % der inhalierten Dosis wird in der Lunge, dem Zielorgan, deponiert. Die verbleibende
Dosis gelangt in den Gastrointestinaltrakt. Ein Teil der unten beschriebenen pharmakokinetischen Ergebnisse wurde mit Dosen erzielt, die über der empfohlenen therapeutischen Dosis liegen.
b) Allgemeine Charakteristika des Wirkstoffs nach Anwendung des Arzneimittels
Resorption: Nach Inhalation durch junge gesunde Probanden zeigten Daten zur Urinausscheidung, dass annähernd 33 % der inhalierten Dosis den systemischen Blutkreislauf erreichen. Orale Tiotropiumbromid-Lösungen weisen eine absolute Bioverfügbarkeit von 2-3 % auf. Ein Einfluss von Nahrungsmitteln auf die Resorption dieser quartären Ammonium-Verbindung ist nicht zu erwarten. Die maximale Tiotropium-Plasmakonzentration wurde 5-7 Minuten nach Inhalation beobachtet. Im Steady State wurde bei COPD-Patienten ein Spitzen-Plasmaspiegel von Tiotropium von 10,5 pg/ml erreicht. Dieser Wert fiel einem Multikompartimentmodell folgend schnell ab. Die tiefste Plasmakonzentration im Steady State betrug 1,60 pg/ml. Bei Asthma-Patienten wurde 5 Minuten nach Verabreichung der gleichen Dosis im Steady State eine maximale Tiotropium-Plasmakonzentration von 5,15 pg/ml erreicht.
Die systemische Exposition nach Tiotropium-Inhalation mit dem Respimat Inhalator oder mit dem HandiHaler Inhalator ist vergleichbar.
Verteilung: Die Plasmaproteinbindung des Wirkstoffes beträgt 72 %, das Verteilungsvolumen 32 l/kg. Örtliche Konzentrationen in der Lunge sind nicht bekannt, jedoch lässt die Anwendungsart wesentlich höhere Werte in der Lunge erwarten. Untersuchungen an Ratten zeigten, dass Tiotropium die BlutHirn-Schranke nicht in einem bedeutenden Maße passiert.
Biotransformation: Das Ausmaß der Metabolisierung ist gering. Dies zeigt sich in der Tatsache, dass 74 % einer intravenösen Dosis bei jungen gesunden Probanden unverändert renal ausgeschieden wird. Der Ester Tiotropiumbromid wird nicht-enzymatisch zu Alkohol (N-Methylscopin) und Säureverbindung (Dithienylglycolsäure) gespalten, die beide an den Muskarinrezeptoren inaktiv sind. In-vitro-Untersuchungen an humanen Lebermikrosomen und humanen Hepatozyten weisen darauf hin, dass ein weiterer Teil des Arzneimittels (< 20 % der Dosis nach intravenöser Anwendung) durch Cytochrom-P450 (CYP)-abhängige Oxidation und anschließende Glutathion-Konjugation zu einer Reihe von Phase-II-Metaboliten metabolisiert wird.
In-vitro-Untersuchungen an Lebermikrosomen zeigen, dass sich der enzymatische Abbauweg durch die CYP-2D6 (und 3A4)-Inhibitoren Chinidin, Ketoconazol und Gestoden hemmen lässt. Somit sind CYP 2D6 und 3A4 an einem Metabolisierungsweg beteiligt, der für die Eliminierung eines geringeren Teils der Dosis verantwortlich ist. Tiotropiumbromid hemmt auch in übertherapeutischen Dosen CYP 1A1, 1A2, 2B6, 2C9, 2C19, 2D6, 2E1 oder 3A in humanen Lebermikrosomen nicht.
Elimination: Die effektive Halbwertszeit von Tiotropium liegt bei gesunden Probanden und COPD-Patienten zwischen 27 und 45 Stunden nach Inhalation. Bei Asthma-Patienten beträgt die effektive Halbwertszeit 34 Stunden. Die Gesamt-Clearance betrug nach intravenöser Anwendung bei jungen gesunden Probanden 880 ml/min. Nach intravenöser Anwendung wird Tiotropium hauptsächlich unverändert mit dem Urin ausgeschieden (74 %). Nach Inhalation der Lösung bis zum Steady State liegt die Urinausscheidung bei COPD-Patienten bei 18,6 % der Dosis (0,93 pg), der Rest besteht hauptsächlich aus nicht resorbiertem Arzneimittel im Darm und wird fäkal ausgeschieden. Nach Inhalation der Lösung durch gesunde Probanden liegt die Urinausscheidung bei 20,1-29,4 % der Dosis, der Rest besteht hauptsächlich aus nicht resorbiertem Arzneimittel im Darm und wird fäkal ausgeschieden. Bei Asthma-Patienten werden im Steady State innerhalb von 24 Stunden nach der Anwendung 11,9 % der Dosis (0,595 pg) unverändert mit dem Urin ausgeschieden. Die Nierenclearance von Tiotropium liegt über der Creatinin-Clearance, was auf eine Ausscheidung in den Urin hinweist.
Nach chronischer einmal täglicher Anwendung bei COPD-Patienten wurde der pharmakokinetische Steady State spätestens nach 7 Tagen erreicht, ohne dass es danach zur Akkumulation kam.
Linearität / Nicht-Linearität: Unabhängig von der Darreichungsform zeigt Tiotropium im therapeutischen Bereich eine lineare Pharmakokinetik.
c) Charakteristika bei Patienten
Ältere Patienten: Wie für alle überwiegend renal ausgeschiedenen Arzneimittel zu erwarten, ging ein fortschreitendes Alter mit einer Abnahme der renalen Tiotropium-Clearance (347 ml/min bei COPD-Patienten < 65 Jahre bis 275 ml/min bei COPD-Patienten > 65 Jahre) einher. Dies führte jedoch nicht zu einem entsprechenden Anstieg der AUC0-6,ss oder Cmax,ss-Werte. Bei Asthma-Patienten ändert sich die Tiotropium-Exposition nicht mit dem Alter.
Patienten mit eingeschränkter Nierenfunktion: Die einmal tägliche Inhalation von Tiotropium bis zum Steady State führte bei COPD-Patienten mit leichter Nierenfunktionsstörung (Creatinin-Clearance 50-80 ml/min) zu leicht erhöhten AUC0_6,ss-Werten (1,8-30 % höher) und ähnlichen Cmax,ss-Werten, verglichen mit COPD-Patienten mit normaler Nierenfunktion (Creatinin-Clearance > 80 ml/min).
Bei COPD-Patienten mit mittlerer bis schwerer Nierenfunktionsstörung (Creatinin-Clearance < 50 ml/min) verdoppelte sich die Gesamtexposition nach intravenöser Applikation einer Einzeldosis von Tiotropium (82 % höhere AUC0-4h und 52 % höhere Cmax), verglichen mit COPD-Patienten mit normaler Nierenfunktion. Dies wurde durch die Plasmakonzentrationen nach Pulverinhalation bestätigt. Bei Asthma-Patienten mit leichter Nierenfunktionsstörung (Creatinin-Clearance 50-80 ml/min) führte die Tiotropium-Inhalation im Vergleich zu Patienten mit normaler Nierenfunktion nicht zu einem relevanten Anstieg der Exposition.
Patienten mit eingeschränkter Leberfunktion: Ein relevanter Einfluss auf die Pharmakokinetik von Tiotropium im Fall einer Leberfunktionsstörung ist nicht zu erwarten. Tiotropium wird hauptsächlich renal ausgeschieden (74 % bei jungen gesunden Probanden) und durch eine einfache nichtenzymatische Esterspaltung zu pharmakologisch inaktiven Produkten abgebaut.
Japanische COPD-Patienten: In einem Studienquervergleich war die mittlere SpitzenPlasmakonzentration von Tiotropium, 10 Minuten nach Inhalation, im Steady State bei japanischen COPD-Patienten 20-70 % höher als bei weißen COPD-Patienten. Es gab jedoch keine Anzeichen für eine höhere Mortalität oder ein höheres kardiales Risiko bei japanischen Patienten, verglichen mit weißen Patienten. Für andere Ethnizitäten oder Rassen sind die pharmakokinetischen Daten unzureichend.
Kinder und Jugendliche:
Im COPD-Studienprogramm gab es keine Kinder und Jugendlichen (siehe Abschnitt 4.2). Im Studienprogramm zur zystischen Fibrose wurden neben Erwachsenen auch Kinder und Jugendliche eingeschlossen.
Bei Zystische-Fibrose-Patienten im Alter von > 5 Jahren lag der Tiotropium-Plasmaspiegel im Steady State, 5 Minuten nach der Inhalation einer 5 Mikrogramm-Dosis, bei 10,1 pg/ml. Er fiel anschließend schnell ab. Der verfügbare Anteil der abgegebenen Dosis war bei Zystische-Fibrose-Patienten im Alter von < 5 Jahren, die einen Spacer und eine Maske benutzten, etwa 3- bis 4-fach geringer als bei Zystische-Fibrose-Patienten im Alter von > 5 Jahren. Bei Zystische-Fibrose-Patienten im Alter von < 5 Jahren bestand ein Zusammenhang zwischen Körpergewicht und Tiotropium-Exposition.
d) Pharmakokinetische/pharmakodynamische Zusammenhänge
Es gibt keinen direkten Zusammenhang zwischen Pharmakokinetik und Pharmakodynamik.
5.3 Präklinische Daten zur Sicherheit
Viele der in den üblichen Studien zur pharmakologischen Sicherheit, zur Toxizität bei wiederholter Anwendung und zur Reproduktionstoxizität beobachteten Wirkungen lassen sich durch die anticholinergen Eigenschaften von Tiotropiumbromid erklären. Bei Tieren wurden typischerweise reduzierte Futteraufnahme, Hemmung der Gewichtszunahme, trockener Mund und Nase, verminderte Tränen- und Speichelsekretion, Mydriasis und Zunahme der Herzfrequenz beobachtet. Weitere wichtige Wirkungen, die in Toxizitätsstudien mit Mehrfachgabe beobachtet wurden, waren: leichte Reizung der Atemwege bei Ratten und Mäusen, die sich in Rhinitis und Epitheländerungen der Nasenhöhle und des Kehlkopfes zeigten, sowie Prostatitis mit proteinreichen Ablagerungen und Lithiasis in der Blase bei Ratten.
Bei jungen Ratten wurden nach Exposition vom 7. Lebenstag bis zur Geschlechtsreife die gleichen direkten und indirekten pharmakologischen Veränderungen beobachtet wie in den Studien zur Toxizität bei wiederholter Anwendung, sowie auch Rhinitis. Es wurde keine systemische Toxizität festgestellt. Es gab keine toxikologisch relevanten Auswirkungen auf die Hauptentwicklungsparameter, auf die Entwicklung der Trachea und der Hauptorgane.
Schädigungen hinsichtlich Schwangerschaft, embryonaler/fetaler Entwicklung, Geburt oder postnataler Entwicklung wurden nur mit maternal toxischen Dosen nachgewiesen. Tiotropiumbromid zeigte bei Ratten oder Kaninchen keine teratogenen Wirkungen.
In einer Studie zur allgemeinen Reproduktion und Fertilität an Ratten ergaben sich bei keiner Dosierung Hinweise auf unerwünschte Effekte auf die Fertilität oder das Paarungsverhalten, weder bei den behandelten Elterntieren noch bei ihren Nachkommen.
Die respiratorischen (Reizungen) und urogenitalen (Prostatitis) Veränderungen sowie reproduktionstoxischen Effekte wurden nach lokalen oder systemischen Dosen beobachtet, die mehr als das 5-fache der therapeutischen Dosis betrugen. Untersuchungen zur Genotoxizität und zum kanzerogenen Potenzial zeigten keine besonderen Gefahren für den Menschen.
6. PHARMAZEUTISCHE ANGABEN
6.1 Liste der sonstigen Bestandteile
Benzalkoniumchlorid Natriumedetat (Ph. Eur.)
Gereinigtes Wasser
Salzsäure 3,6 % (zur pH-Einstellung)
6.2 Inkompatibilitäten
Nicht zutreffend.
6.3 Dauer der Haltbarkeit
3 Jahre
Haltbarkeit nach Anbruch: 3 Monate
6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Nicht einfrieren!
6.5 Art und Inhalt des Behältnisses
Art und Material des Behältnisses im Kontakt mit dem Arzneimittel:
Lösung, gefüllt in eine Polyethylen/Polypropylen-Patrone mit einer Schutzkappe aus Polypropylen mit integriertem Silikondichtungsring. Die Patrone befindet sich in einem Aluminiumzylinder.
Packungsgrößen und beigefügtes Medizinprodukt:
• Einzelpackung:
• Doppelpackung:
• Dreifachpackung:
• Klinikpackung:
1 Respimat Inhalator und 1 Patrone; entspricht 60 Hüben (30 therapeutische Dosen)
2 Einzelpackungen, bestehend aus je 1 Respimat Inhalator und 1 Patrone, entspricht 60 Hüben (30 therapeutische Dosen)
3 Einzelpackungen, bestehend aus je 1 Respimat Inhalator und 1 Patrone, entspricht 60 Hüben (30 therapeutische Dosen)
8 Einzelpackungen, bestehend aus je 1 Respimat Inhalator und 1 Patrone, entspricht 60 Hüben (30 therapeutische Dosen)
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
7. INHABER DER ZULASSUNG
Boehringer Ingelheim International GmbH Binger Str. 173 55216 Ingelheim am Rhein Deutschland
8. ZULASSUNGSNUMMER
52523.00.01
9. DATUM DER ERTEILUNG DER ZULASSUNG / VERLÄNGERUNG DER ZULASSUNG
Datum der Erteilung der Zulassung: 12.09.2007
Datum der letzten Verlängerung der Zulassung: 09.10.2012
10. STAND DER INFORMATION
Februar 2016
11. WEITERE ANGABEN
Verkaufsabgrenzung
Verschreibungspflichtig
Örtlicher Vertreter des Zulassungsinhabers in Deutschland Boehringer Ingelheim Pharma GmbH & Co. KG Binger Str. 173 55216 Ingelheim am Rhein
Tel.: 0 800 / 77 90 900 Fax: 0 61 32 / 72 99 99 E-Mail: info@boehringer-ingelheim.de
Seite 20 von 20