Urapidil Stragen I.v. 25 Mg Injektionslösung
1. BEZEICHNUNG DES ARZNEIMITTELS
Urapidil Stragen i.v. 25 mg Injektionslösung
2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
1 ml enthält 5 mg Urapidil.
5 ml Ampulle enthält 25 mg Urapidil.
Vollständige Auflistung der sonstigen Bestandteile siehe Abschnitt 6.1.
3. DARREICHUNGSFORM
Injektionslösung, die auch für Infusionszwecke verdünnt werden kann.
Klare, farblose Lösung mit einem pH-Wert von 5,6 bis 6,6.
Ohne sichtbare Partikel.
4. KLINISCHE ANGABEN
4.1 Anwendungsgebiete
Bluthochdrucknotfälle (z.B. ein kritischer Blutdruckanstieg), schwere und sehr schwere Formen der Bluthochdruckkrankheit oder behandlungsresistenter Bluthochdruck.
Zur kontrollierten Senkung des Blutdrucks bei Patienten mit Bluthochdruck während und/oder nach einer Operation.
4.2 Dosierung und Art der Anwendung
Dosierung
Für Bluthochdrucknotfälle, schwere und sehr schwere Formen des Bluthochdrucks und behandlungsresistenten Bluthochdruck
Intravenöse Injektion
10 - 50 mg Urapidil wird langsam mittels einer intravenösen Injektion verabreicht, dabei wird der Blutdruck ständig überwacht. Ein Abfall des Blutdrucks kann innerhalb von 5 Minuten nach Verabreichung der Injektion erwartet werden.
Die Injektion von 10 - 50 mg Urapidil kann wiederholt werden, je nachdem wie der Blutdruck reagiert.
Eine intravenöse Infusion oder eine Applikation via Spritzenpumpe werden zur Erhaltung des Blutdruckwerts verwendet, der durch die Injektion erzielt wurde.
Hinweise zur Verdünnung des Arzneimittels vor der Anwendung, siehe Abschnitt 6.6.
Die kompatible Maximalmenge ist 4 mg Urapidil pro ml Infusionslösung.
Applikationsrate: Die Infusionsrate richtet sich nach dem Blutdruck der betreffenden Person. Anfänglich empfohlene maximale Infusionsrate: 2 mg/Min.
Erhaltungsdosis: Durchschnittlich 9 mg/Std. Werden 250 mg Urapidil (50 ml) zu 500 ml Infusionslösung zugegeben, entspricht 1 mg = 44 Tropfen = 2,2 ml.
Die kontrollierte Senkung des Blutdrucks, wenn der Blutdruck während und/oder nach einer Operation erhöht ist.
Intravenöse Infusion oder Applikation mittels Spritzenpumpe werden zur Erhaltung des Blutdruckwerts verwendet, die durch die Injektion erzielt wurden.
Dosierungsplan
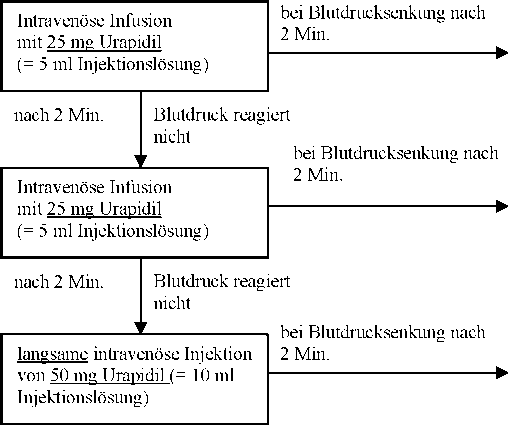
Blutdruck durch
Infusion
stabilisiert
Anfänglich bis zu 6 mg in 1 - 2 Min., dann reduzieren
Art der Anwendung
Hinweis
Urapidil Stragen i.v. wird auf dem Rücken liegenden Patienten intravenös als Injektion oder Infusion verabreicht.
Die Dosis kann in einer oder mehreren Injektionen oder als langsame intravenöse Infusion verabreicht werden. Injektionen können mit einer anschließenden langsamen Infusion kombiniert werden.
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von intravenösem Urapidil ist bei Kindern im Alter von 0 - 18 Jahren bisher noch nicht erwiesen.
Eine Dosierungsempfehlung kann daher nicht gegeben werden.
Ältere Patienten
Bei älteren Patienten sind blutdrucksenkende Mittel mit angemessener Vorsicht zu verabreichen und anfangs in kleinen Dosen, da bei diesen Patienten häufig eine veränderte Empfindlichkeit gegenüber dieser Art von Präparaten auftritt.
Patienten mit Störungen der Nieren- und/oder Leberfunktion
Bei Patienten mit Nieren- und/oder Leberfunktionsstörungen muss möglicherweise die Dosis Urapidil reduziert werden.
Behandlungsdauer
Eine Behandlungsdauer von 7 Tagen wurde aus toxikologischer Sicht als sicher befunden; bei parenteralen blutdrucksenkenden Mitteln sollte dieser Zeitraum generell nicht überschritten werden. Die erneute parenterale Behandlung ist möglich, wenn der Blutdruck wieder steigt.
Es ist möglich eine akute parenterale Therapie mit wechselnder bis fortgesetzter Behandlung mit oralen blutdrucksenkenden Mitteln zu überlappen.
4.3 Gegenanzeigen
Urapidil Stragen i.v. sollte nicht verwendet werden, wenn eine Überempfindlichkeit (Allergie) gegen den Wirkstoff oder einen der Inhaltsstoffe vorliegt. Urapidil Stragen i.v. sollte in Fällen mit Aortenisthmusstenose und arteriovenösem Shunt (ausgenommen hämodynamisch nicht aktive Dialyse-Shunts) nicht verwendet werden.
4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Vorsichtsmaßnahmen für die Anwendung
- Bei Herzinsuffizienz, die von einer Beeinträchtigung der mechanischen Funktion wie beispielsweise Stenose der Aorten- oder Mitralklappen, pulmonaler Embolie oder eingeschränkter Herztätigkeit infolge einer perikardialen Krankheit hervorgerufen wurde;
- Bei Patienten mit Leberfunktionsstörungen;
- Bei Patienten mit moderaten bis schweren Nierenfunktionsstörungen;
- Bei älteren Patienten;
- Bei Patienten, die gleichzeitig Cimetidin erhalten (siehe Abschnitt 4.5 Wechselwirkung mit anderen Arzneimitteln und andere Formen der Wechselwirkung).
Wenn Urapidil nicht als blutdrucksenkendes Mittel zur Erstbehandlung gegeben wird, muss ausreichend Zeit verstreichen, damit die Wirkung des/der zuvor verabreichten blutdrucksenkenden Mittel(s) beobachtet werden kann. Die gewählte Dosis Urapidil sollte entsprechend niedriger sein.
Ein zu schneller Abfall des Blutdrucks kann zu Bradykardie oder Herzstillstand führen.
Infolge des Gehalts an Propylenglykol können bei der Verabreichung von Urapidil Stragen i.v. Symptome beobachtet werden, die der Wirkung von Alkohol vergleichbar sind.
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Dosis, d.h. es ist so gut wie natriumfrei.
4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Die blutdrucksenkende Wirkung von Urapidil kann durch die gleichzeitige Verabreichung von AlphaRezeptor-Blockern, wie u.a. jene, die für urologische Krankheiten gegeben werden, Vasodilatotoren und andere blutdrucksenkende Arzneimittel und bei Krankheiten, die mit Hypovolämie einhergehen können (Durchfall, Erbrechen), und Alkohol verstärkt werden.
Die Kombination von Urapidil mit Baclofen ist nur mit Vorsicht in Erwägung zu ziehen, da Baclofen die blutdrucksenkende Wirkung steigern kann.
Gleichzeitig verabreichtes Cimetidin hemmt den Urapidil-Metabolismus. Die Urapidil-Serumkonzentration erhöht sich wahrscheinlich um 15%. Daher sollte eine Dosisreduktion erwogen werden.
Folgende gleichzeitige Verabreichung bedürfen der besonderen Aufmerksamkeit:
- Imipramin (blutdrucksenkende Wirkung und Risiko von orthostatischer Hypotonie)
- Neuroleptika (blutdrucksenkende Wirkung und Risiko von orthostatischer Hypotonie) und
- Kortikoide (Reduktion der blutdrucksenkenden Wirkung durch Wasser-Natrium-Retention)
Da es noch keine adäquaten Erfahrungswerte über die Kombinationsbehandlung mit ACE-Hemmern gibt, wird diese derzeit nicht empfohlen.
4.6 Fertilität, Schwangerschaft und Stillzeit
Schwangerschaft
Urapidil Stragen i.v. wird während der Schwangerschaft nicht empfohlen. Es liegen keine adäquaten Daten zur Verwendung von Urapidil bei schwangeren Frauen vor.
Tierexperimentelle Studien haben eine Reproduktionstoxizität ohne Teratogenität gezeigt (Kapitel 5.3). Aufgrund der Einschränkungen der Studien sind die potenziellen Risiken für Menschen nicht bekannt.
Stillzeit
Da zur Ausscheidung in die Muttermilch keine Daten vorliegen, wird das Stillen im Falle einer Behandlung mit Urapidil nicht empfohlen.
4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Das Arzneimittel hat einen geringen Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
Die Reaktion auf die Behandlung kann von Patient zu Patient unterschiedlich sein. Das gilt insbesondere bei Behandlungsbeginn, nach Änderungen der Behandlung oder bei gleichzeitigem Alkoholkonsum.
4.8 Nebenwirkungen
Zusammenfassung des Sicherheitsprofils
In den meisten Fällen können die folgenden Nebenwirkungen auf einen zu rapiden Absturz des Blutdrucks zurückgeführt werden. Die Erfahrung hat jedoch gezeigt, dass diese innerhalb von Minuten wieder abklingen, auch bei langsamer Infusion. Daher sollte über eine Behandlungsunterbrechung je nach dem Schweregrad der Nebenwirkung entschieden werden.
Tabellarische Auflistung der Nebenwirkungen
Nachfolgend sind die Nebenwirkungen nach MedDRA-System-Organklassen aufgelistet.
Bei den Häufigkeitsangaben werden folgende Kategorien zugrunde gelegt:
Sehr häufig: >1/10 Häufig: >1/100 bis <1/10 Gelegentlich: >1/1.000 bis <1/100 Selten: >1/10.000 bis <1/1.000
Sehr selten: <1/10.000, einschließlich isolierte Meldung
Nicht bekannt (kann anhand der verfügbaren Daten nicht bewertet werden).
|
Häufigkeit System Organklasse |
Sehr häufig (>1/10) |
Häufig (>1/100 bis <1/10) |
Gelegentlich (>1/1.000 bis < 1/100) |
Selten (> 1/10.000 bis < 1/1.1000 |
Sehr selten (>1/10.000) |
Nicht bekannt (kann anhand der verfügbaren Daten nicht bewertet werden) |
|
Störungen des Blut- und Lymphsystems |
Thrombozyto penie | |||||
|
Herzkrankheiten |
Palpitationen; Tachykardie; Bradykardie; Druckgefühl auf Brust; Atemnot; Herzrhythmusstör ungen | |||||
|
Erkrankungen des Gastrointestinal- |
Übelkeit |
Erbrechen |
|
trakts | ||||||
|
Allgemeine Erkrankungen und Probleme an der Verabreichungsstelle |
Müdigkeit |
Asthenie | ||||
|
Erkrankungen des Nervensystems |
Schwindel, Kopf schmerzen | |||||
|
Psychiatrische Erkrankungen |
Ruhelosigkeit | |||||
|
Erkrankungen der Geschlechtsorgane und der Brustdrüse |
Priapismus | |||||
|
Erkrankungen der Atemwege, des Brustraums und Mediastinums |
Verstopfte Nase | |||||
|
Erkrankungen der Haut und des Unterhautzellgewebes |
Schweißaus brüche |
Symptome kutaner allergischer Reaktionen (Pruritus, Ausschläge, Exanthem) |
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Arzneimittel und Medizinprodukte, Abt. Pharmakovigilanz, Kurt-Georg-Kiesinger-Allee 3, D-53175 Bonn, Website: www.bfarm.de anzuzeigen.
4.9 Überdosierung
Symptome
Symptome einer Überdosis sind Schwindel, orthostatische Hypotonie und Kollaps sowie Müdigkeit und reduziertes Reaktionsvermögen.
Behandlung einer Überdosis
Ein übermäßiger Abfall des Blutdrucks kann durch Anheben der Beine und Volumentherapie behoben werden. Wenn diese Maßnahmen nicht ausreichen, können blutgefäßverengende Präparate langsam intravenös injiziert werden, während der Blutdruck überwacht wird. In sehr seltenen Fällen ist die Verabreichung von Catecholaminen (d.h. Adrenalin, 0,5 - 1,0 mg mit isotoner Natriumchloridlösung auf 10 ml verdünnt) erforderlich.
5. PHARMAKOLOGISCHE EIGENSCHAFTEN
5.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: ANTIADRENERGIKA, PERIPHER WIRKENDE, Alpha-Adrenorezeptor-Antagonisten, ATC-Code: C02CA06
6
Urapidil führt zu einem Abfall des systolischen und diastolischen Blutdrucks, indem die periphere Resistenz gesenkt wird.
Die Herzfrequenz bleibt weitgehend konstant.
Die Herzleistung wird nicht verändert; die Herzleistung, die infolge eines erhöhten peripheren Widerstands reduziert ist, kann sich erhöhen.
Wirkungsmechanismus
Urapidil hat sowohl zentrale als auch periphere Wirkungen.
• Peripher: Urapidil blockiert vor allem postsynaptische Alpha-Rezeptoren und hemmt demzufolge die vasokonstriktive Wirkung von Catecholaminen.
• Zentral: Urapidil hat zudem eine zentrale Wirkung. Es modulierte die Aktivität der Gehirnzentren, die das Kreislaufsystem regeln. Daher wird die reaktive Erhöhung des Sympatikotonus gehemmt oder der Sympatikotonus wird reduziert.
5.2 Pharmakokinetische Eigenschaften
Nach der intravenösen Verabreichung von 25 mg Urapidil, ist die Serumkonzentration (anfängliche Distributionsphase, terminale Ausscheidungsphase) biphasisch. Die Distributionsphase hat eine Halbwertzeit von ungefähr 35 Min. Das Distributionsvolumen beträgt 0,8 (0,6 - 1,2) l/kg.
Urapidil wird hauptsächlich in der Leber metabolisiert. Der Hauptmetabolit ist Urapidil, das an der 4. Position des Phenylkerns hydroxyliert ist, welcher keine bemerkenswerte blutdrucksenkende Wirkung aufweist. Der Metabolit von O-demethyliertem Urapidil weist ungefähr dieselbe biologische Aktivität wie Urapidil auf, entsteht aber in einem viel geringeren Umfang.
50 - 70% von Urapidil und seinen Metaboliten wird in Menschen über die Nieren ausgeschieden, wie auch ungefähr 15% der als pharmakologisch aktives Urapidil verabreichten Dosis; der Rest wird fäkal als Metaboliten ausgeschieden, primär als para-hydroxyliertes Urapidil, das den Blutdruck nicht senkt. Nach einer intravenösen Bolusinjektion wurde die Halbwertzeit im Serum mit 2,7 (1,8 - 3,9) Std. bestimmt.
Die Plasmaproteinbindung von Urapidil (humanes Serum) liegt bei 80% in-vitro. Diese relativ geringe Plasmaproteinbindung von Urapidil könnte erklären, warum bis heute keine Wechselwirkungen zwischen Urapidil und Arzneimitteln bekannt sind, die stark an Plasmaprotein gebunden sind.
Bei fortgeschrittener Leber- und/oder Niereninsuffizienz und bei älteren Patienten ist das Distributionsvolumen und die Ausscheidung von Urapidil verringert und die AusscheidungsHalbwertzeit verlängert.
Urapidil penetriert die Blut-Hirn-Schranke und ist placentagängig.
5.3 Präklinische Daten zur Sicherheit
Akute Toxizität
Zur akuten Toxizität wurden Untersuchungen mit Urapidilhydrochlorid an Mäusen und Ratten durchgeführt. Die LD50-Werte (bezogen auf Urapidil-Base) liegen nach oraler Gabe zwischen 508 und 750 mg/kg KG und nach intravenöser Applikation zwischen 140 und 260 mg/kg KG. Toxische Wirkungen wurden vor allem als Sedierung, Ptose, reduzierte Motilität, Verlust des Schutzreflexes und Hypothermie, Schnappatmung, Zyanose, Tremor und Konvulsionen vor dem Tod beobachtet.
Chronische Toxizität /Subchronische Toxizität
Studien zur chronischen Toxizität wurden an Ratten nach oraler Gabe mit dem Futter über 6 und 12 Monate mit Dosierungen bis zu 250 mg/kg KG/ Tag durchgeführt. Beobachtet wurden Sedierung, Ptosis, verminderte Körpergewichtszunahme, eine Verlängerung des Oestruszyklus und verminderte Uterusgewichte.
Am Hund wurde die chronische Toxizität in Studien über 6 und 12 Monate mit Dosierungen bis zu 64 mg/kg KG geprüft. Dosierungen ab 30 mg/kg KG/Tag verursachten Sedierung, Hypersalivation und Tremor. Klinische oder histopathologische Veränderungen wurden am Hund nicht festgestellt.
Mutagenes und tumorinduzierendes Potenzial
In Bakterienstudien (AMES-Test, host-vermittelte Assay), Untersuchungen bei humanen Lymphozyten und dem Knochenmark-Metaphase-Test bei Mäusen zeigte Urapidil keine mutagenen Merkmale. Ein Test auf DNS-Reparatur an Rattenhepatozyten verlief negativ.
Aus Kanzerogenitätsuntersuchungen an Mäusen und Ratten über 18 und 24 Monate haben sich keine für den Menschen relevanten Hinweise auf ein tumorerzeugendes Potential ergeben. In speziellen Untersuchungen an Ratten und Mäusen zeigte sich, dass Urapidil den Prolaktinspiegel erhöht. Beim Nager führt ein erhöhter Prolaktinspiegel zur Stimulation des Wachstums von Mammagewebe. Aufgrund der Kenntnisse über den Wirkungsmechanismus ist diese Wirkung für den Menschen bei therapeutischer Dosierung nicht zu erwarten und konnte in klinischen Studien nicht nachgewiesen werden.
Reproduktionstoxizität
Studien zur Reproduktionstoxizität in Ratten, Mäusen und Kaninchen ergeben keinen Hinweis auf eine teratogene Wirkung.
Studien bei Ratten und Kaninchen ergaben eine Reproduktionstoxizität von Urapidil. Die Nebenwirkungen bestanden aus einer reduzierten Trächtigkeitsrate bei Ratten; einer reduzierten Gewichtszunahme und Futter- und Wasseraufnahme bei trächtigen Kaninchen; einer reduzierten Lebendfötusrate bei Kaninchen; und einer reduzierten perinatalen Überlebensrate und Gewichtszunahme bei neugeborenen Ratten.
Die Reproduktionsstudie zeigte, dass der Östruszyklus von weiblichen Ratten verlängert war, was die Studie für chronische Toxizität auch gezeigt hatte. Diese Wirkung, wie z. B. das reduzierte Uterusgewicht beim chronischen Test, wird dem erhöhten Prolactin-Spiegel zugeschrieben, der bei Nagetieren nach der Behandlung mit Urapidil auftritt. Die Fertilität von Weibchen wurde nicht beeinträchtigt.
Im Hinblick auf die erheblichen Unterschiede zwischen den Gattungen können diese Ergebnisse jedoch nicht als anwendbar auf Menschen erachtet werden. In langfristigen klinischen Studien konnte keine Wirkung auf das Hypophysen- Gonaden-System bei der Frau gezeigt werden.
6. PHARMAZEUTISCHE ANGABEN
6.1 Liste der sonstigen Bestandteile
Propylenglykol,
Natriumdihydrogenphosphat-Dihydrat,
Salzsäure (37% w/w),
Natriummonohydrogenphosphat-Dihydrat (Ph.Eur.),
Salzsäure (3,7% w/w),
Natriumhydroxid-Lösung (4% w/w),
Wasser für Injektionszwecke.
6.2 Inkompatibilitäten
Das Arzneimittel darf, außer mit den unter Abschnitt 6.6 aufgeführten, nicht mit anderen Arzneimitteln gemischt werden.
Die folgenden Wirkstoffe (oder Lösung zur Rekonstitution/Verdünnung) sollten nicht gleichzeitig verabreicht werden:
Alkalische Injektions- und Infusionslösungen Dies kann zu Trübung oder Ausflockungen führen.
04891
6.3 Dauer der Haltbarkeit 3 Jahre.
Nach der ersten Öffnung/Verdünnung:
Die chemische und physikalische Stabilität wurde für 50 Stunden bei 15-25 °C nachgewiesen.
Aus mikrobiologischen Gesichtspunkten sollte das Arzneimittel umgehend verwendet werden.
Wenn die gebrauchsfertige Zubereitung nicht sofort eingesetzt wird, ist der Anwender für die Dauer und die Bedingungen der Aufbewahrung verantwortlich. Sofern die Herstellung der gebrauchsfertigen Zubereitung nicht unter kontrollierten und validierten Bedingungen erfolgt, darf diese normalerweise nicht länger als 24 Stunden bei 2 - 8 °C aufbewahrt werden.
6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Nicht über 30°C lagern.
Aufbewahrungsbedingungen nach Verdünnung des Arzneimittels, siehe Abschnitt 6.3.
6.5 Art und Inhalt des Behältnisses
Ampullen aus klarem Glas (Typ I Ph. Eur.)
Packungsgröße: 5 Ampullen
6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung
Die 100 mg Ampulle darf nur zur Stabilisierung des Blutdrucks per Infusion verwendet werden.
Für die anfängliche Behandlung stehen Ampullen mit 25 mg und 50 mg Urapidil zur Verfügung.
Diese Dosierungsstärken können nach der Verdünnung auch für die intravenöse Infusion verwendet werden.
Die Verdünnung wird unter aseptischen Bedingungen hergestellt.
Die Lösung ist vor der Verabreichung visuell auf Schwebstoffteilchen und Verfärbung zu prüfen. Nur klare und farblose Lösung verwenden.
Herstellung von verdünnter Lösung:
- Intravenöse Infusion:
250 mg Urapidil (2 Ampullen 100 mg Urapidil + 1 Ampulle 50 mg Urapidil) in 500 ml eines der kompatiblen Lösungsmittel geben.
- Spritzenpumpe:
100 mg Urapidil wird in eine Spritzenpumpe aufgezogen und mit einem der kompatiblen Lösungsmittel auf ein Volumen von 50 ml verdünnt.
Kompatible Lösungsmittel zur Verdünnung:
- Natriumchlorid 9 mg/ml (0,9%) Infusionslösung
- Glukose 50 mg/ml (5%)
- Glukose 100 mg/ml (10%)
Nur zum einmaligen Gebrauch.
Nicht verwendete Lösungen oder Abfallmaterial sind, entsprechend den nationalen Anforderungen zu beseitigen.
7. INHABER DER ZULASSUNG
Stragen Nordic A/S DK-3400 Hillerod Dänemark
Mitvertrieb:
STRAGEN Pharma GmbH Eupener Straße 135-137 D-50933 Köln Deutschland
8. ZUL ASSUN GSNUMMER(N)
75114.00.00
9. DATUM DER ERTEILUNG DER ZULASSUNG/VERLÄNGERUNG DER ZULASSUNG
04/Mai/2012
10. STAND DER INFORMATION
31.08.2015
04891
10