Valganciclovir-Ratiopharm 450 Mg Filmtabletten
FACHINFORMATION
1. BEZEICHNUNG DES ARZNEIMITTELS
Valganciclovir-ratiopharm® 450 mg Filmtabletten
2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
Jede Tablette enthält 496,3 mg Valganciclovirhydrochlorid, entsprechend 450 mg Valganciclovir. Sonstiger Bestandteil mit bekannter Wirkung:
Jede Filmtablette enthält 6,365 mg Lactose (als Lactose-Monohydrat).
Vollständige Auflistung der sonstigen Bestandteile siehe Abschnitt 6.1.
3. DARREICHUNGSFORM
Filmtablette
Rosafarbene, ovale Filmtabletten mit abgeschrägten Kanten und der Prägung „93“ auf der einen Seite und „5465“ auf der anderen Seite.
4. KLINISCHE ANGABEN
4.1 Anwendungsgebiete
Valganciclovir-ratiopharm® ist zur Initial- und Erhaltungstherapie der Cytomegalievirus(CMV)-Retinitis bei Patienten mit erworbenem Immundefekt-Syndrom (AIDS) angezeigt.
Valganciclovir-ratiopharm® ist zur Prophylaxe einer CMV-Erkrankung bei CMV-negativen Patienten angezeigt, die ein Organtransplantat von einem CMV-positiven Spender erhalten haben.
4.2 Dosierung und Art der Anwendung
Dosierung
Vorsicht: Um eine Überdosierung zu vermeiden, müssen die Dosierungsempfehlungen strikt eingehalten werden (siehe Abschnitte 4.4 und 4.9).
Valganciclovir wird nach oraler Anwendung rasch und umfassend zu Ganciclovir metabolisiert. Oral angewendetes Valganciclovir 900 mg zweimal täglich entspricht therapeutisch einer intravenösen Gabe von Ganciclovir 5 mg/kg zweimal täglich.
Standarddosierung bei Erwachsenen
Initialtherapie der CMV-Retinitis:
Bei Patienten mit aktiver CMV-Retinitis beträgt die empfohlene Dosis 900 mg Valganciclovir (zwei Valganciclovir-ratiopharm® 450 mg Tabletten) zweimal täglich über 21 Tage, möglichst mit einer Mahlzeit eingenommen. Eine länger dauernde Initialtherapie kann das Risiko myelotoxischer Wirkungen erhöhen (siehe Abschnitt 4.4).
Erhaltungstherapie der CMV-Retinitis:
Nach der Initialtherapie bzw. bei Patienten mit inaktiver CMV-Retinitis beträgt die empfohlene Dosis 900 mg Valganciclovir (zwei Valganciclovir-ratiopharm® 450 mg Tabletten) einmal täglich, möglichst mit einer Mahlzeit eingenommen. Bei Verschlechterung der Retinitis kann die Initialtherapie wiederholt werden; es ist jedoch an die Möglichkeit einer viralen Arzneimittelresistenz zu denken.
Prophylaxe der CMV-Erkrankung nach Organtransplantation:
Bei Patienten nach einer Nierentransplantation beträgt die empfohlene Dosis 900 mg (zwei Valganciclovir-ratiopharm® 450 mg Tabletten) einmal täglich. Die Behandlung wird innerhalb von 10 Tagen nach der Transplantation begonnen und bis 100 Tage nach der Transplantation fortgeführt. Die Prophylaxe kann bis 200 Tage nach der Transplantation fortgeführt werden (siehe Abschnitte 4.4, 4.8 und 5.1).
Bei Patienten nach einer anderen Organtransplantation als einer Nierentransplantation beträgt die empfohlene Dosis 900 mg (zwei Valganciclovir-ratiopharm® 450 mg Tabletten) einmal täglich. Die Behandlung wird innerhalb von 10 Tagen nach der Transplantation begonnen und bis 100 Tage nach der Transplantation fortgeführt.
Die Tabletten werden möglichst mit einer Mahlzeit eingenommen.
Besondere Dosierungshinweise
Patienten mit eingeschränkter Nierenfunktion:
Der Serumkreatininspiegel oder die Kreatininclearance müssen sorgfältig überwacht werden. Die Dosis ist in Abhängigkeit von der Kreatininclearance entsprechend der folgenden Tabelle anzupassen (siehe Abschnitte 4.4 und 5.2).
Die geschätzte Kreatininclearance (CrCl [ml/min]) kann entsprechend dem gemessenen Serumkreatinin nach folgender Formel berechnet werden:
Für Männer = (140 - Alter [ Jahrei) x (Körpergewicht Tkgl)
(72) x (0,011 x Serumkreatinin [^mol/l])
Für Frauen = 0,85 x Wert für Männer
|
CrCl (ml/min) |
Initialdosis Valganciclovir |
Erhaltungs- bzw. Prophylaxedosis Valganciclovir |
|
> 60 |
900 mg (2 Tabletten) zweimal täglich |
900 mg (2 Tabletten) einmal täglich |
|
40 - 59 |
450 mg (1 Tablette) zweimal täglich |
450 mg (1 Tablette) einmal täglich |
|
25 - 39 |
450 mg (1 Tablette) einmal täglich |
450 mg (1 Tablette) alle 2 Tage |
|
10 - 24 |
450 mg (1 Tablette) alle 2 Tage |
450 mg (1 Tablette) zweimal wöchentlich |
|
< 10 |
nicht empfohlen |
nicht empfohlen |
Dialysepflichtige Patienten:
Für dialysepflichtige Patienten (CrCl < 10 ml/min) kann keine Dosierungsempfehlung gegeben werden. Daher soll Valganciclovir-ratiopharm® bei diesen Patienten nicht angewendet werden (siehe Abschnitte 4.4 und 5.2).
Patienten mit eingeschränkter Leberfunktion:
Die Sicherheit und Wirksamkeit von Valganciclovir bei Patienten mit eingeschränkter Leberfunktion wurden nicht untersucht (siehe Abschnitt 5.2).
Kinder und Jugendliche:
Die Sicherheit und Wirksamkeit von Valganciclovir bei Kindern und Jugendlichen ist bisher nicht durch adäquate und entsprechend kontrollierte Studien erwiesen. Die aktuell verfügbaren Daten sind in den Abschnitten 4.8, 5.1 und 5.2 dargestellt, eine Dosierungsempfehlung kann jedoch nicht gegeben werden.
Ältere Patienten:
Bei dieser Patientengruppe wurden Sicherheit und Wirksamkeit nicht untersucht.
Patienten mit schwerer Leukopenie, Neutropenie, Anämie, Thrombozytopenie und Panzytopenie:
Vor Therapieeinleitung siehe Abschnitt 4.4.
Wenn es unter der Therapie mit Valganciclovir-ratiopharm® zu einer deutlichen Verschlechterung des Blutbildes kommt, ist eine Behandlung mit hämatopoetischen Wachstumsfaktoren und/oder eine Therapieunterbrechung in Betracht zu ziehen (siehe Abschnitte 4.4 und 4.8).
Art der Anwendung
Valganciclovir-ratiopharm® wird oral angewendet und soll möglichst mit einer Mahlzeit eingenommen werden (siehe Abschnitt 5.2).
Vorsichtsmaßnahmen vor/bei der Handhabung bzw. vor/während der Anwendung des Arzneimittels Die Tabletten dürfen nicht zerbrochen oder zerdrückt werden. Da Valganciclovir-ratiopharm® beim Menschen potenziell teratogen und karzinogen wirkt, ist beim Umgang mit zerbrochenen Tabletten Vorsicht geboten (siehe Abschnitt 4.4). Direkter Kontakt von zerbrochenen oder zerdrückten Tabletten mit Haut oder Schleimhäuten ist zu vermeiden. Wenn es dennoch zu einem solchen Kontakt kommt, ist die Berührungsstelle gründlich mit Wasser und Seife zu reinigen, die Augen sind mit sterilem Wasser oder, wenn dies nicht zur Verfügung steht, mit Leitungswasser gründlich zu spülen.
4.3 Gegenanzeigen
Valganciclovir-ratiopharm® darf bei Patienten mit Überempfindlichkeit gegen Valganciclovir, Ganciclovir oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile nicht angewendet werden.
Wegen der ähnlichen chemischen Struktur von Valganciclovir-ratiopharm® und Aciclovir sowie Valaciclovir ist eine Kreuzallergie zwischen diesen Wirkstoffen möglich. Valganciclovir-ratiopharm® darf daher bei Patienten mit Überempfindlichkeit gegenüber Aciclovir und Valaciclovir nicht angewendet werden.
Valganciclovir-ratiopharm® darf nicht während der Stillzeit angewendet werden (siehe Abschnitt 4.6).
4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Vor Beginn der Behandlung mit Valganciclovir sind die Patienten hinsichtlich des potenziellen Risikos für den Fetus aufzuklären. In Tierstudien erwies sich Ganciclovir als mutagen, teratogen, aspermatogen sowie karzinogen und beeinträchtigte die weibliche Fertilität. Valganciclovir-ratiopharm® ist daher beim Menschen als potenziell teratogen und karzinogen anzusehen und verursacht möglicherweise angeborene
Fehlbildungen und Krebserkrankungen (siehe Abschnitt 5.3). Ferner gilt es als wahrscheinlich, dass Valganciclovir zu einer vorübergehenden oder dauerhaften Unterdrückung der Spermatogenese führt.
Frauen im gebärfähigen Alter sind anzuweisen, während der Behandlung eine wirksame Empfängnisverhütung zu praktizieren. Männer müssen darauf hingewiesen werden, unter der Behandlung und noch mindestens 90 Tage danach Kondome zur Kontrazeption zu benutzen, es sei denn, bei ihrer Partnerin ist die Möglichkeit einer Schwangerschaft ausgeschlossen (siehe Abschnitte 4.6, 4.8 und 5.3).
Valganciclovir hat langfristig ein karzinogenes und reproduktionstoxisches Potenzial.
Unter Behandlung mit Valganciclovir (und Ganciclovir) wurden schwere Leukopenien, Neutropenien, Anämien, Thrombozytopenien, Panzytopenien, Knochenmarkdepressionen und aplastische Anämien beobachtet. Die Behandlung sollte nicht begonnen werden, wenn die absolute Neutrophilenzahl unter 500 Zellen/gl oder die Thrombozytenzahl unter 25.000/gl oder der Hämoglobinspiegel unter 8 g/dl liegt (siehe Abschnitte 4.2 und 4.8).
Wird die Prophylaxe länger als 100 Tage weitergeführt, sollte das mögliche Risiko zur Entwicklung einer Leukopenie und Neutropenie in Betracht gezogen werden (siehe Abschnitte 4.2, 4.8 und 5.1).
Bei der Anwendung von Valganciclovir-ratiopharm® bei Patienten mit bestehender Zytopenie oder einer arzneimittelbedingten Zytopenie in der Anamnese sowie bei Patienten unter Strahlenbehandlung ist Vorsicht geboten.
Es wird empfohlen, während der Behandlung das Differenzialblutbild und die Thrombozytenzahl zu überwachen. Bei Patienten mit eingeschränkter Nierenfunktion kann eine verstärkte hämatologische Überwachung angezeigt sein. Wenn sich eine schwere Leukopenie, Neutropenie, Anämie und/oder Thrombozytopenie entwickelt, wird empfohlen, eine Behandlung mit hämatopoetischen Wachstumsfaktoren und/oder eine Therapieunterbrechung in Betracht zu ziehen (siehe Abschnitte 4.2 und 4.8).
Die Bioverfügbarkeit von Ganciclovir aus einer Einzeldosis von 900 mg Valganciclovir beträgt etwa 60 %, verglichen mit etwa 6 % nach oraler Anwendung von 1000 mg Ganciclovir (in Form von Kapseln). Überdosierungen mit Ganciclovir können lebensbedrohliche Nebenwirkungen hervorrufen. Deshalb ist bei Einleitung der Behandlung und bei Umstellung von der Initial- auf die Erhaltungstherapie eine sorgfältige Einhaltung der Dosierungsempfehlungen angebracht. Eine Umstellung von einer oralen Behandlung mit Ganciclovir (in Form von Kapseln) auf Valganciclovir in Form von Valganciclovir-ratiopharm® ist nicht im Verhältnis von 1:1 möglich. Patienten, die von Ganciclovir-Kapseln umgestellt werden, sind auf das Risiko einer Überdosierung hinzuweisen, wenn sie mehr als die vorgeschriebene Anzahl Valganciclovir-ratiopharm® Tabletten einnehmen (siehe Abschnitte 4.2 und 4.9).
Bei Patienten mit eingeschränkter Nierenfunktion sind Dosisanpassungen auf Basis der Kreatininclearance erforderlich (siehe Abschnitte 4.2 und 5.2).
Bei Dialysepatienten soll Valganciclovir-ratiopharm® nicht angewendet werden (siehe Abschnitte 4.2 und 5.2).
Bei Patienten unter Imipenem/Cilastatin und Ganciclovir wurden Krampfanfälle beobachtet. Valganciclovir-ratiopharm® darf nur dann gleichzeitig mit Imipenem/Cilastatin angewendet werden, wenn der Nutzen für den Patienten die möglichen Risiken überwiegt (siehe Abschnitt 4.5).
Bei Anwendung von Valganciclovir-ratiopharm® zusammen mit (a) Didanosin, (b) Arzneimitteln, die bekanntermaßen myelosuppressiv sind (z. B. Zidovudin) oder (c) Stoffen, die die Nierenfunktion beeinträchtigen, sind die Patienten engmaschig auf Anzeichen zusätzlicher toxischer Wirkungen zu überwachen (siehe Abschnitt 4.5).
In die kontrollierte klinische Studie mit Valganciclovir zur Prophylaxe der CMV-Erkrankung nach Organtransplantation, die in Abschnitt 5.1 näher beschrieben wird, waren keine Patienten nach Lungen- oder Darmtransplantation eingeschlossen. Die Erfahrungen in dieser Patientengruppe sind deshalb begrenzt.
Valganciclovir-ratiopharm® enthält Lactose. Patienten mit den seltenen, heriditären Problemen einer Galactose-Intoleranz, eines Lapp-Lactasemangels oder einer Glucose-Galactose-Malabsorption dürfen dieses Arzneimittel nicht einnehmen.
4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Wechselwirkungen mit Valganciclovir
In vivo wurden keine Studien zur Erfassung von Wechselwirkungen mit Valganciclovir durchgeführt. Da Valganciclovir rasch und umfassend zu Ganciclovir metabolisiert wird, werden für Valganciclovir die gleichen Wechselwirkungen wie bei Ganciclovir erwartet.
Wirkungen anderer Arzneimittel auf Ganciclovir
Imipenem/Cilastatin
Bei gleichzeitiger Einnahme von Ganciclovir und Imipenem/Cilastatin wurden Krampfanfälle beobachtet. Diese Arzneimittel dürfen nur dann gleichzeitig gegeben werden, wenn der Nutzen für den Patienten die möglichen Risiken überwiegt (siehe Abschnitt 4.4).
Probenecid
Die gleichzeitige Anwendung von Probenecid und oralem Ganciclovir führte zu einer statistisch signifikant reduzierten renalen Clearance von Ganciclovir (20 %) und somit zu einer statistisch signifikant höheren Exposition (40 %). Diese Veränderungen entsprechen einem Interaktionsmechanismus, bei dem die Substanzen um die renale tubuläre Sekretion konkurrieren. Deshalb sollen Patienten, die Probenecid und Valganciclovir-ratiopharm® einnehmen, engmaschig auf toxische Wirkungen von Ganciclovir überwacht werden.
Wirkungen von Ganciclovir auf andere Arzneimittel
Zidovudin
Wurde Zidovudin zusammen mit Ganciclovir eingenommen, so trat eine kleine (17 %), aber statistisch signifikante Erhöhung des AUC-Wertes von Zidovudin auf. Ferner wurde bei gemeinsamer Gabe mit Zidovudin eine - wenn auch nicht statistisch signifikante - Tendenz zu niedrigeren Ganciclovir-Konzentrationen beobachtet. Da jedoch sowohl Zidovudin als auch Ganciclovir potenziell Neutropenie und Anämie auslösen können, führt eine Kombinationstherapie in voller Dosishöhe unter Umständen bei einigen Patienten zu Unverträglichkeiten (siehe Abschnitt 4.4).
Didanosin
Die Plasmakonzentrationen von Didanosin waren bei gleichzeitiger Anwendung von Ganciclovir (sowohl intravenös als auch oral) durchgängig erhöht. Bei oralen Ganciclovir-Dosen von 3 und 6 g/Tag wurde ein Anstieg der AUC von Didanosin um 84 bis 124 % beobachtet. Ebenso wurde bei intravenösen Dosen von 5 und 10 mg/kg/Tag ein Anstieg der AUC von Didanosin um 38 bis 67 % beobachtet. Klinisch signifikante Auswirkungen auf die Ganciclovir-Konzentration traten nicht auf. Die Patienten sind engmaschig auf toxische Wirkungen von Didanosin hin zu überwachen (siehe Abschnitt 4.4).
Mycophenolatmofetil
Aufgrund der Ergebnisse einer Studie mit einer Einzelgabe der empfohlenen oralen Dosis Mycophenolatmofetil (MMF) und intravenös verabreichtem Ganciclovir sowie der bekannten Auswirkungen einer Nierenfunktionsstörung auf die Pharmakokinetik von MMF und Ganciclovir wird erwartet, dass die gleichzeitige Gabe dieser Substanzen (die um die renale tubuläre Sekretion konkurrieren können) zu einem Anstieg der Konzentrationen des phenolischen Glucuronids von Mycophenolsäure (MPAG) und Ganciclovir führt. Es werden keine wesentlichen Veränderungen der Pharmakokinetik von Mycophenolsäure (MPA) erwartet und eine Dosisanpassung von MMF ist daher nicht erforderlich. Bei Patienten mit eingeschränkter Nierenfunktion, die gleichzeitig mit MMF und Ganciclovir behandelt werden, ist die empfohlene Ganciclovir-Dosis einzuhalten und die Patienten sind sorgfältig zu überwachen. Da sowohl MMF als auch Ganciclovir potenziell Neutropenie und Leukopenie verursachen können, sind die Patienten hinsichtlich additiver Toxizitäten zu überwachen.
Stavudin
Bei gleichzeitiger Anwendung von Stavudin und oralem Ganciclovir wurden keine klinisch signifikanten Wechselwirkungen beobachtet.
Trimethoprim
Bei gleichzeitiger Gabe von Trimethoprim und oralem Ganciclovir wurden keine klinisch signifikanten pharmakokinetischen Wechselwirkungen beobachtet. Es besteht jedoch die Möglichkeit verstärkter Toxizität, da beide Substanzen bekanntermaßen myelosuppressiv sind. Daher sollen die beiden Arzneimittel nur dann gleichzeitig angewendet werden, wenn der mögliche Nutzen die Risiken überwiegt.
Andere antiretroviral wirksame Substanzen
Bei Anwendung therapeutisch wirksamer Konzentrationen ist weder eine synergistische noch eine antagonistische Wirkung auf die Hemmung von HIV (in Anwesenheit von Ganciclovir) oder CMV (in Anwesenheit einer Reihe antiretroviral wirksamer Substanzen) wahrscheinlich. Stoffwechselinteraktionen, zum Beispiel mit Proteaseinhibitoren und nicht nukleosidischen Reverse-Transkriptase-Inhibitoren (NNRTIs), sind unwahrscheinlich, da Cytochrom-P450 weder am Valganciclovir- noch am Ganciclovir-Metabolismus beteiligt ist.
Andere mögliche Arzneimittelwechselwirkungen
Es besteht die Möglichkeit einer erhöhten toxischen Wirkung, wenn Valganciclovir gleichzeitig mit oder unmittelbar vor bzw. nach anderen Arzneimitteln gegeben wird, welche die Replikation von sich rasch teilenden Zellpopulationen hemmen, wie sie im Knochenmark, den Hoden und den Keimschichten der Haut und der Magen-Darm-Schleimhaut vorkommen. Beispiele für solche Arzneimittel sind Dapson, Pentamidin, Flucytosin, Vincristin, Vinblastin, Adriamycin, Amphotericin B, Trimethoprim/Sulfonamid-Kombinationen, Nukleosidanaloga und Hydroxyharnstoff.
Da Ganciclovir über die Nieren ausgeschieden wird (Abschnitt 5.2), kann die Toxizität auch verstärkt werden, wenn Valganciclovir gleichzeitig mit Arzneimitteln angewendet wird, die die renale Clearance von Ganciclovir verringern und damit dessen Konzentration im Körper erhöhen könnten. Die renale Clearance von Ganciclovir könnte durch zwei Mechanismen gehemmt werden: (a) durch Nephrotoxizität, verursacht durch Wirkstoffe wie z. B. Cidofovir und Foscarnet, und (b) durch kompetitive Hemmung der aktiven tubulären Sekretion in der Niere, verursacht z. B. durch andere Nukleosidanaloga.
Deshalb sollen alle diese Arzneimittel nur dann zusammen mit Valganciclovir angewendet werden, wenn der mögliche Nutzen die möglichen Risiken überwiegt (siehe Abschnitt 4.4).
4.6 Fertilität, Schwangerschaft und Stillzeit
Schwangerschaft
Es liegen keine Daten zur Anwendung von Valganciclovir bei Schwangeren vor. Ganciclovir, der aktive Metabolit von Valganciclovir, passiert die menschliche Plazenta. Aufgrund seines pharmakologischen Wirkungsmechanismus und der in tierexperimentellen Studien mit Ganciclovir beobachteten Reproduktionstoxizität (siehe Abschnitt 5.3) besteht grundsätzlich das Risiko teratogener Wirkungen beim Menschen.
Valganciclovir-ratiopharm® darf während der Schwangerschaft nur dann angewendet werden, wenn der therapeutische Nutzen für die Mutter das potenzielle teratogene Risiko für das Kind überwiegt.
Stillzeit
Es ist nicht bekannt, ob Ganciclovir in die Muttermilch übergeht. Die Möglichkeit, dass Ganciclovir in die Muttermilch übergeht und bei gestillten Säuglingen schwerwiegende Nebenwirkungen auslöst, kann jedoch nicht ausgeschlossen werden. Daher muss abgestillt werden (siehe Abschnitt 4.3).
Fertilität
Frauen im gebärfähigen Alter sind anzuweisen, während der Behandlung wirksame Empfängnisverhütungsmaßnahmen zu treffen. Männer müssen darauf hingewiesen werden, während der Behandlung mit Valganciclovir-ratiopharm® und für mindestens 90 Tage danach Kondome zur Kontrazeption zu benutzen, es sei denn, bei ihrer Partnerin ist die Möglichkeit einer Schwangerschaft ausgeschlossen (siehe Abschnitt 5.3).
4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Es wurden keine Studien zu den Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen durchgeführt.
Bei Anwendung von Valganciclovir-ratiopharm® und/oder Ganciclovir wurden Krampfanfälle, Sedierung, Schwindel, Ataxie und/oder Verwirrtheitszustände berichtet. Solche Wirkungen können Aufgaben, die Aufmerksamkeit erfordern, beeinträchtigen, darunter auch die Verkehrstüchtigkeit des Patienten und die Fähigkeit zum Bedienen von Maschinen.
4.8 Nebenwirkungen
Valganciclovir ist ein Prodrug von Ganciclovir, das nach Einnahme rasch und umfassend zu Ganciclovir metabolisiert wird. Die für Ganciclovir bekannten Nebenwirkungen können daher auch bei der Anwendung von Valganciclovir erwartet werden. Alle Nebenwirkungen, die bei Valganciclovir in klinischen Studien beobachtet wurden, sind zuvor schon mit Ganciclovir beobachtet worden. Die bei Erwachsenen am häufigsten berichteten Nebenwirkungen nach Anwendung von Valganciclovir sind Neutropenie, Anämie und Diarrhö.
Das Risiko für das Auftreten einer Diarrhö ist bei Valganciclovir höher als bei intravenös angewendetem Ganciclovir. Darüber hinaus ist das Risiko für das Auftreten einer Neutropenie und Leukopenie bei Valganciclovir höher als bei eingenommenem Ganciclovir.
Bei Patienten mit CMV-Retinitis, die mit Valganciclovir behandelt werden, wird häufiger eine schwere Neutropenie (< 500 neutrophile Zellen/pl) beobachtet als bei Patienten nach einer Organtransplantation, die Valganciclovir erhalten.
In unten stehender Tabelle sind die Häufigkeiten der Nebenwirkungen angeführt, die in klinischen Studien mit Valganciclovir, eingenommenem Ganciclovir oder Ganciclovir i.v. berichtet wurden. Die genannten Nebenwirkungen wurden in klinischen Studien zur Initial- oder Erhaltungstherapie der CMV-Retinitis bei Patienten mit AIDS bzw. zur Prophylaxe einer CMV-Erkrankung bei Patienten nach Leber-, Nieren- oder Herztransplantation beobachtet. Der Klammerausdruck („schwer“) in der Tabelle weist darauf hin, dass die genannte Nebenwirkung sowohl in leichter bzw. mäßiger Intensität als auch in schwerwiegender bzw. lebensbedrohlicher Intensität bei den Patienten auftrat.
Innerhalb jeder Häufigkeitsgruppe werden die Nebenwirkungen nach abnehmendem Schweregrad angegeben.
|
Körpersystem |
Sehr häufig |
Häufig |
Gelegentlich |
Selten |
|
(> 1/10) |
(> 1/100 bis < 1/10) |
(> 1/1000 bis < 1/100) |
(> 1/10.000 bis < 1/1000) | |
|
Infektionen und parasitäre Erkrankungen |
Orale Candidiasis, Sepsis (Bakteriämie, Virämie), Zellulitis, Harnwegsinfektionen | |||
|
Erkrankungen des Blutes und des Lymphsystems |
(Schwere) Neutropenie, Anämie |
Schwere Anämie, (schwere) Thrombozytopenie, (schwere) Leukopenie, (schwere) Panzytopenie |
Knochenmark versagen |
Aplastische Anämie |
|
Erkrankungen des Immunsystems |
Anaphylaktische Reaktion | |||
|
Stoffwechsel- und Ernährungsstörungen |
Appetitabnahme, Anorexie | |||
|
Psychiatrische Erkrankungen |
Depression, Angst, Verwirrtheit, Veränderungen des Denkens |
Agitiertheit, psychotische Zustände, Halluzinationen | ||
|
Erkrankungen des Nervensystems |
Kopfschmerzen, Insomnie, Dysgeusie (Geschmacksstörungen), Hypästhesie, Parästhesie, periphere Neuropathie, Schwindelgefühl, Krampfanfall |
Tremor | ||
|
Augenerkrankungen |
Makulaödem, Netzhautablösung, Mouches volantes, Augenschmerzen |
Sehstörungen, Konjunktivitis | ||
|
Erkrankungen des Ohrs und des Labyrinths |
Ohrenschmerzen |
Taubheit | ||
|
Herzerkrankungen |
Arrhythmie | |||
|
Gefäßerkrankungen |
Hypotonie | |||
|
Erkrankungen der Atemwege, des |
Dyspnö |
Husten |
|
Brustraums und Mediastinums | ||||
|
Erkrankungen des Gastrointestinaltrakts |
Diarrhö |
Übelkeit, Erbrechen, Abdominalschmerzen, Oberbauchschmerzen, Dyspepsie, Obstipation, Flatulenz, Dysphagie |
Aufgetriebenes Abdomen, Mundgeschwür, Pankreatitis | |
|
Leber- und Gallenerkrankungen |
(Schwere) Störungen der Leberfunktion, erhöhte Werte für die alkalische Phosphatase im Blut, Aspartat- Aminotransferase erhöht |
Alanin- Aminotransferase erhöht | ||
|
Erkrankungen der Haut und des Unterhautzellgewebes |
Dermatitis, nächtliche Schweißausbrüche, Pruritus |
Alopezie, Urtikaria, trockene Haut | ||
|
Skelettmuskulatur-, Bindegewebs-und Knochenerkrankunge n |
Rückenschmerzen, Myalgie, Arthralgie, Muskelspasmen | |||
|
Erkrankungen der Nieren und Harnwege |
Renale Kreatininclearance reduziert, Nierenfunktionsstörunge n |
Hämaturie, Nierenversagen | ||
|
Erkrankungen der Geschlechtsorgane und der Brustdrüse |
Männliche Infertilität | |||
|
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort |
Müdigkeit, Fieber, Schüttelfrost, Schmerzen, Schmerzen im Brustraum, allgemeines Krankheitsgefühl, Asthenie | |||
|
Untersuchungen |
Gewichtsabnahme, erhöhte Blutkreatininwerte |
Schwere Thrombozytopenien können mit möglicherweise lebensbedrohlichen Blutungen verbunden sein. Kinder und Jugendliche
Es existieren nur sehr begrenzte Daten zur Behandlung von Kindern mit Valganciclovir (siehe Abschnitte 5.1 und 5.2). Die folgende Tabelle bietet eine Übersicht mit allen Nebenwirkungen, die bei mehr als 10 % (sehr häufig) der insgesamt behandelten Kindern aufgetreten sind:
|
Körpersystem |
Sehr häufige Nebenwirkungen in klinischen Studien |
|
Erkrankungen des Blutes und des Lymphsystems |
Anämie, Neutropenie |
|
Gefäßerkrankungen |
Hypertonie |
|
Erkrankungen der Atemwege, des Brustraums und Mediastinums |
Infektionen der oberen Atemwege |
|
Erkrankungen des Gastrointestinaltrakts |
Diarrhö, Übelkeit, Erbrechen und Obstipation |
|
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort |
Fieber, Transplantatabstoßung |
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Arzneimittel und Medizinprodukte, Abt. Pharmakovigilanz, Kurt-Georg-Kiesinger-Allee 3, D-53175 Bonn, Website: www.bfarm.de anzuzeigen.
4.9 Überdosierung
Erfahrungen mit Überdosierungen von Valganciclovir
Bei einem Erwachsenen entwickelte sich nach mehreren Tagen unter einer Dosierung, die mindestens um das 10-fache höher lag als die entsprechend dem Grad der Niereninsuffizienz des Patienten (verminderte Kreatininclearance) empfohlene Dosierung, eine letale Knochenmarkdepression (medulläre Aplasie).
Es ist zu erwarten, dass eine Überdosierung von Valganciclovir möglicherweise auch zu einer stärkeren Nierentoxizität führen kann (siehe Abschnitte 4.2 und 4.4).
Hämodialyse und Flüssigkeitszufuhr können zur Senkung der Blutplasmaspiegel nach einer Überdosis Valganciclovir sinnvoll sein (siehe Abschnitt 5.2).
Erfahrungen mit Überdosierung von intravenös angewendetem Ganciclovir
Aus klinischen Studien und während der Erfahrung nach Markt-Einführung sind Berichte von Überdosierungen mit i.v. Ganciclovir eingegangen. In einigen dieser Fälle wurden keine unerwünschten Ereignisse angegeben. Bei den meisten Patienten traten eines oder mehrere der folgenden unerwünschten Ereignisse auf:
- Hämatotoxizität: Panzytopenie, Knochenmarkdepression, medulläre Aplasie, Leukopenie, Neutropenie, Granulozytopenie.
- Hepatotoxizität: Hepatitis, Leberfunktionsstörung.
- Nephrotoxizität: Verschlechterung einer Hämaturie bei einem Patienten mit bereits bestehender Nierenfunktionsstörung, akutes Nierenversagen, Kreatininanstieg.
- Gastrointestinale Toxizität: Bauchschmerzen, Diarrhö, Erbrechen.
- Neurotoxizität: generalisierter Tremor, Krampfanfall.
5. PHARMAKOLOGISCHE EIGENSCHAFTEN
5.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: ATC-Code: J05AB14 (Antiinfektiva zur systemischen Anwendung, antivirale Arzneimittel zur systemischen Anwendung, direkt wirkende antivirale Arzneimittel).
Wirkmechanismus:
Valganciclovir ist ein L-Valinester (Prodrug) von Ganciclovir. Nach oraler Anwendung wird Valganciclovir rasch und umfassend von den intestinalen und hepatischen Esterasen zu Ganciclovir metabolisiert. Ganciclovir ist ein synthetisches Analogon von 2'-Desoxyguanosin und hemmt sowohl in vitro als auch in vivo die Replikation von Herpesviren. Zu den empfindlichen Viren beim Menschen gehören das humane Cytomegalievirus (HCMV), die Herpes-simplex-Viren 1 und 2 (HSV-1 und HSV-2), die humanen Herpesviren 6, 7 und 8 (HHV-6, HHV-7, HHV-8), das Epstein-Barr-Virus (EBV), das Varicella-ZosterVirus (VZV) und das Hepatitis-B-Virus (HBV).
In CMV-infizierten Zellen wird Ganciclovir zuerst von der viruseigenen Proteinkinase pUL97 zu Ganciclovirmonophosphat phosphoryliert. Eine weitere Phosphorylierung erfolgt durch zelluläre Kinasen zu Ganciclovirtriphosphat, das dann im Zellinnern nur langsam metabolisiert wird. Die Halbwertszeit von Ganciclovirtriphosphat in HSV- und HCMV-infizierten Zellen beträgt nach Entzug des extrazellulären Ganciclovirs 18 bzw. 6 bis 24 Stunden. Da die Phosphorylierung größtenteils von der viralen Kinase abhängt, erfolgt die Phosphorylierung von Ganciclovir vorzugsweise in virusinfizierten Zellen.
Die virostatische Aktivität von Ganciclovir basiert auf der Hemmung der viralen DNA-Synthese durch: (a) kompetitive Hemmung des Einbaus von Desoxyguanosintriphosphat in die DNA durch die virale DNA-Polymerase und (b) Einbau von Ganciclovirtriphosphat in die virale DNA mit nachfolgendem Abbruch der viralen DNA-Elongation oder starker Einschränkung der weiteren viralen DNA-Elongation.
Antivirale Aktivität
Die antivirale Aktivität in vitro, gemessen als IC50 von Ganciclovir gegenüber CMV, liegt im Bereich von 0,08 pM (0,02 pg/ml) bis 14 pM (3,5 pg/ml).
Die klinische antivirale Wirkung von Valganciclovir wurde bei der Behandlung von AIDS-Patienten mit neu diagnostizierter CMV-Retinitis nachgewiesen (klinische Studie WV15376). Die CMV-Ausscheidung im Urin nahm ab von 46 % (32/69) der Patienten bei Studienbeginn auf 7 % (4/55) der Patienten nach vierwöchiger Behandlung mit Valganciclovir.
Klinische Wirksamkeit
Behandlung der CMV-Retinitis:
In einer Studie wurden Patienten mit neu diagnostizierter CMV-Retinitis randomisiert einer Initialtherapie mit entweder Valganciclovir 900 mg zweimal täglich oder i.v. Ganciclovir 5 mg/kg zweimal täglich zugewiesen. Der Anteil der Patienten mit photographisch erfasstem Fortschreiten der CMV-Retinitis in der vierten Woche war in beiden Behandlungsgruppen vergleichbar, und zwar lag der Anteil an Patienten mit fortschreitender Erkrankung bei 7/70 bzw. 7/71 in den Behandlungsarmen mit i.v. Ganciclovir bzw. Valganciclovir.
Nach der Initialtherapie erhielten alle Patienten in dieser Studie eine Erhaltungstherapie mit Valganciclovir in einer Dosierung von 900 mg täglich. Die mittlere (mediane) Zeit von der Randomisierung bis zur
Progression der CMV-Retinitis betrug in der Gruppe, die die Initial- und Erhaltungstherapie mit Valganciclovir erhielt, 226 (160) Tage und in der Gruppe, die eine Initialtherapie mit intravenösem Ganciclovir und eine Erhaltungstherapie mit Valganciclovir erhielt, 219 (125) Tage.
Prophylaxe einer CMV-Erkrankung nach Transplantation:
Bei Patienten mit hohem CMV-Erkrankungsrisiko (D+/R-) nach Herz-, Leber- oder Nierentransplantation wurde eine doppelblinde, double-dummy kontrollierte klinische Studie mit wirkstoffhaltigem Vergleichspräparat durchgeführt, bei der die Patienten entweder Valganciclovir (900 mg einmal täglich) oder Ganciclovir oral (1000 mg dreimal täglich) erhielten, beginnend innerhalb von 10 Tagen und bis zum Tag 100 nach der Transplantation. Patienten nach Lungen- oder Darmtransplantation waren nicht in die Studie eingeschlossen. Die Inzidenz der CMV-Erkrankung (CMV-Syndrom plus gewebsinvasive Erkrankung) betrug während der ersten 6 Monate nach Transplantation 12,1 % im Studienarm, der Valganciclovir erhielt (n = 239), verglichen mit 15,2 % im Studienarm, der Ganciclovir oral erhielt (n = 125). Die überwiegende Mehrzahl der Fälle trat nach Beendigung der Prophylaxe auf (d. h. nach Tag 100), wobei die Fälle im Arm mit Valganciclovir durchschnittlich später auftraten als diejenigen im Studienarm mit oralem Ganciclovir. Die Inzidenz akuter Abstoßungsreaktionen in den ersten 6 Monaten betrug 29,7 % bei Patienten, die randomisiert Valganciclovir erhielten, gegenüber 36,0 % bei Patienten, die Ganciclovir oral erhielten. Die Inzidenz eines Transplantatverlustes war mit 0,8 % in beiden Studienarmen gleich.
Eine doppelblinde, placebokontrollierte Studie wurde an 326 Nierentransplantationspatienten mit hohem CMV-Erkrankungsrisiko (D+/R-) durchgeführt, um die Wirksamkeit und Sicherheit einer von 100 auf 200 Tage nach der Transplantation verlängerten CMV-Prophylaxe mit Valganciclovir zu bewerten. Die Patienten wurden randomisiert (1:1) und erhielten Valganciclovir Tabletten (900 mg einmal täglich) beginnend innerhalb von 10 Tagen nach der Transplantation, entweder bis zum Tag 200 nach der Transplantation oder bis zum Tag 100 nach der Transplantation, gefolgt von einer Placebogabe für weitere 100 Tage.
Der Anteil der Patienten, die eine CMV-Erkrankung während der ersten 12 Monate nach der Transplantation entwickelten, wird in nachstehender Tabelle gezeigt.
Prozentualer Anteil der Nierentransplantationspatienten mit einer CMV-Erkrankung1, 12 Monate ITT-PopulationA
|
Valganciclovir 900 mg einmal täglich 100 Tage (n =163) |
Valganciclovir 900 mg einmal täglich 200 Tage (n =155) |
Differenz zwischen den Behandlungsgruppen | |
|
Patienten mit bestätigter |
71 (43,6 %) |
36 (23,2 %) |
20,3 % |
|
oder Verdacht auf CMV- Erkrankung2 |
[35,8 %; 51,5 %] |
[16,8 %; 30,7 %] |
[9,9 %; 30,8 %] |
|
Patienten mit bestätigter |
60 (36,8 %) |
25 (16,1 %) |
20,7 % |
|
CMV-Erkrankung |
[29,4 %; 44,7 %] |
[10,7 %; 22,9 %] |
[10,9 %; 30,4 %] |
1 CMV-Erkrankung ist definiert entweder als CMV-Syndrom oder Gewebe-invasive CMV. 2 Eine bestätigte CMV ist ein klinisch bestätigter Fall einer CMV-Erkrankung. Eine CMV-Erkrankung wurde angenommen, wenn es in Woche 52 keine Bewertung und keine Bestätigung einer CMV-Erkrankung vor diesem Zeitpunkt gab.
A Die Ergebnisse nach 24 Monaten stehen in Einklang mit den Ergebnissen nach 12 Monaten: Die bestätigten oder angenommenen CMV-Erkrankungen betrugen 48,5 % im Studienarm über die Behandlung von 100 Tagen und 34,2 % im Studienarm über die Behandlung von 200 Tagen. Die Differenz zwischen beiden Behandlungsarmen betrug 14,3 % [3,2 %; 25,3 %].
Es haben signifikant weniger Hochrisikopatienten nach einer Nierentransplantation eine CMV-Erkrankung entwickelt, nachdem sie Valganciclovir zur CMV-Prophylaxe 200 Tage lang nach der Transplantation erhalten haben, im Vergleich zu Patienten, die Valganciclovir zur CMV-Prophylaxe bis Tag 100 nach der Transplantation erhalten haben.
Die Überlebensrate des Transplantats sowie das Auftreten einer durch Biopsie nachgewiesenen akuten Abstoßungsreaktion waren in beiden Behandlungsarmen vergleichbar. Die Überlebensrate des Transplantats betrug 12 Monate nach der Transplantation 98,2 % (160/163) bei dem 100-Tage-Dosierungsschema und
98,1 % (152/155) bei dem 200-Tage-Dosierungsschema. Bis zu 24 Monate nach der Transplantation sind vier weitere Fälle von Transplantatverlust aufgetreten, die alle in der Behandlungsgruppe mit einer Behandlungsdauer von 100 Tagen aufgetreten sind. Die Inzidenz der durch Biopsie nachgewiesenen akuten Abstoßungsreaktion betrug 12 Monate nach der Transplantation 17,2 % (28/163) bei dem 100-Tage-Dosierungsschema und 11,0 % (17/155) bei dem 200-Tage-Dosierungsschema. Bis zu 24 Monate nach der Transplantation wurde von einem weiteren Fall in der Behandlungsgruppe mit einer Behandlungsdauer von 200 Tagen berichtet.
Virusresistenz
Nach chronischer Anwendung von Valganciclovir können gegen Ganciclovir resistente Viren auftreten, indem es zu einer Selektion von Mutationen im Gen der viralen Kinase (UL97), das für die Monophosphorylierung von Ganciclovir verantwortlich ist, und/oder im viralen Gen der Polymerase (UL54) kommt. Viren, die Mutationen im UL97-Gen enthalten, sind nur gegen Ganciclovir resistent, während Viren mit Mutationen im UL54-Gen resistent gegen Ganciclovir sind, jedoch eine Kreuzresistenz auch gegen andere Virostatika, die ebenfalls an der viralen Polymerase angreifen, aufweisen können.
Behandlung der CMV-Retinitis:
Die genotypische Analyse von CMV-Isolaten in polymorphkernigen Leukozyten von 148 Patienten mit CMV-Retinitis, die an einer klinischen Studie teilnahmen, zeigte, dass nach 3, 6, 12 bzw. 18 Monaten Behandlung mit Valganciclovir 2,2 %, 6,5 %, 12,8 % bzw. 15,3 % der Isolate UL97-Mutationen enthalten.
Prophylaxe einer CMV-Erkrankung nach Transplantation:
Studie mit wirkstoffhaltigem Vergleichsarzneimittel
Die Virusresistenz wurde durch genotypische Analyse von CMV in Proben polymorphkerniger Leukozyten untersucht, die am Tag 100 (d. h. am Ende der Prophylaxe mit der Studienmedikation) und, bei Verdacht auf eine CMV-Erkrankung, bis zu 6 Monate nach Transplantation genommen wurden. Von 245 Patienten, die für die Behandlung mit Valganciclovir randomisiert waren, standen 198 Tag-100-Proben für die Untersuchung zur Verfügung. Es wurden keine Resistenzmutationen bezüglich Ganciclovir beobachtet. Demgegenüber wurden in den 103 Proben der Patienten aus dem Vergleichsarm, die Ganciclovir oral erhielten, 2 Ganciclovir-Resistenzmutationen (1,9 %) festgestellt.
Von den 245 Patienten, die für Valganciclovir randomisiert waren, wurden Proben von 50 Patienten mit Verdacht auf CMV-Erkrankung untersucht, wobei wiederum keine Resistenzmutationen gefunden wurden. Von den 127 Patienten, die dem Ganciclovir-Vergleichsarm zugeteilt waren, wurden Proben von 29 Patienten mit Verdacht auf CMV-Erkrankung untersucht, wobei zwei Resistenzmutationen festgestellt wurden, entsprechend einer Inzidenz von 6,9 %.
Studie zur Verlängerung der Prophylaxe von 100 auf 200 Tage nach Transplantation Genotypische Analysen wurden beim UL54- und UL97-Gen durchgeführt in extrahiertem Virus von je 72 Patienten. Die Patienten haben den Resistenzanalysekriterien entsprochen: Patienten mit positiver Viruslast (> 600 Kopien/ml) am Ende der Behandlung zur Prophylaxe und/oder Patienten, die bis zu 12 Monate (52 Wochen) nach der Transplantation eine bestätigte CMV-Erkrankung entwickelt haben. Drei Patienten in jeder Behandlungsgruppe hatten eine bestätigte Ganciclovir-Resistenzmutation.
Kinder und Jugendliche
In einer Phase-II-Studie zur Untersuchung der Pharmakokinetik und Sicherheit bei pädiatrischen Empfängern eines Organtransplantats (im Alter von 4 Monaten bis 16 Jahren, n = 63) wurde Valganciclovir einmal täglich bis zu 100 Tage lang angewendet. Die Dosierung erfolgte mittels eines Algorithmus, der bei Kindern zu einer vergleichbaren Exposition wie bei Erwachsenen führte (siehe Abschnitt 5.2). Die Beobachtung der Patienten nach der Behandlung erstreckte sich auf 12 Wochen. Der CMV D/R-Ausgangsserostatus war: D+/R- bei 40 %, D+/R+ bei 38 %, D-/R+ bei 19 % und D-/R- bei 3 % der Patienten. Das CM-Virus wurde bei 7 Patienten nachgewiesen. Die beobachteten Nebenwirkungen des Arzneimittels waren ähnlich denen bei Erwachsenen (siehe Abschnitt 4.8). Diese Daten sind zu eingeschränkt, um daraus Schlüsse über die Wirksamkeit oder Dosierungsempfehlungen für pädiatrische Patienten ableiten zu können.
Die Pharmakokinetik und Sicherheit von in Einzeldosen angewendetem Valganciclovir (Dosisbereich 14-1620 mg/kg/Dosis) wurde bei 24 Neugeborenen (im Alter von 8-34 Tagen) untersucht, die unter einer symptomatischen, kongenitalen CMV-Erkrankung litten (siehe Abschnitt 5.2). Die Neugeborenen erhielten eine antivirale Therapie über 6 Wochen, wobei 19 der 24 Patienten bis zu 4 Wochen mit oral angewendetem Valganciclovir und anschließend 2 Wochen mit i.v. angewendetem Ganciclovir behandelt wurden. Die verbleibenden 5 Patienten erhielten während der Studie die meiste Zeit i.v. verabreichtes Ganciclovir. Diese Therapie wird derzeit nicht für Valganciclovir empfohlen. Sowohl das Studiendesign als auch die erhaltenen Ergebnisse sind zu eingeschränkt, um angemessene Schlussfolgerungen zur Wirksamkeit und Sicherheit von Valganciclovir zu ziehen.
5.2 Pharmakokinetische Eigenschaften
Die pharmakokinetischen Eigenschaften von Valganciclovir wurden bei HIV- und CMV-seropositiven Patienten, bei Patienten mit AIDS und CMV-Retinitis sowie bei Patienten nach einer Organtransplantation untersucht.
Resorption
Valganciclovir ist ein Prodrug von Ganciclovir. Es wird aus dem Magen-Darm-Trakt gut resorbiert und in der Darmwand und Leber rasch und umfassend zu Ganciclovir metabolisiert. Die systemische Verfügbarkeit von Valganciclovir ist vorübergehend und gering. Die absolute Bioverfügbarkeit von Ganciclovir aus Valganciclovir beträgt bei allen untersuchten Patientenpopulationen etwa 60 %, und die resultierende Verfügbarkeit von Ganciclovir ist vergleichbar mit der Exposition nach intravenöser Verabreichung von Ganciclovir (siehe unten). Im Vergleich dazu beträgt die Bioverfügbarkeit von Ganciclovir nach Gabe von 1000 mg Ganciclovir oral (als Kapseln) 6-8 %.
Valganciclovir bei HlV-positiven und CMV-positiven Patienten:
Die systemische Exposition bei HIV-positiven und CMV-positiven Patienten nach zweimal täglicher Anwendung von Ganciclovir und Valganciclovir über eine Woche zeigt die folgenden Werte:
|
Parameter |
Ganciclovir (5 mg/kg, i.v.) n = 18 |
Valganciclovir (900 mg, p.o.) n = 25 | |
|
Ganciclovir |
Valganciclovir | ||
|
AUC(0-12 h) (Mg X h/ml) |
28,6 ± 9,0 |
32,8 ± 10,1 |
0,37 ± 0,22 |
|
Cmax (Mg/ml) |
10,4 ± 4,9 |
6,7 ± 2,1 |
0,18 ± 0,06 |
Es wurde gezeigt, dass die Wirksamkeit von Ganciclovir hinsichtlich der Verlängerung der Zeit bis zum Fortschreiten der CMV-Retinitis mit der systemischen Exposition (AUC) korreliert.
Valganciclovir bei Patienten nach Organtransplantation:
Die systemische Ganciclovir-Exposition im Steady State bei Patienten nach einer Organtransplantation nach täglicher oraler Gabe von Ganciclovir und Valganciclovir zeigt die folgenden Werte:
|
Parameter |
Ganciclovir (1000 mg 3 x tägl.) n = 82 |
Valganciclovir (900 mg, 1 x tägl.) n = 161 |
|
Ganciclovir | ||
|
AUC(0-24 h) (gg x h/ml) |
28,0 ± 10,9 |
46,3 ± 15,2 |
|
Cmax (gg/ml) |
1,4 ± 0,5 |
5,3 ± 1,5 |
Nach Einnahme von Valganciclovir gemäß dem Nierenfunktions-Dosierungsalgorithmus ist die systemische Exposition von Ganciclovir bei Herz-, Nieren- und Lebertransplantat-Empfängern ähnlich.
Einfluss von Nahrung:
Eine Dosisproportionalität hinsichtlich des AUC-Wertes von Ganciclovir nach Anwendung von Valganciclovir in einem Dosisbereich von 450 bis 2625 mg konnte nur nach Nahrungsaufnahme nachgewiesen werden. Wurde Valganciclovir zusammen mit einer Mahlzeit in der empfohlenen Dosis von 900 mg gegeben, so wurden für Ganciclovir sowohl höhere mittlere AUC-Werte (etwa 30 %) als auch höhere mittlere Cmax-Werte (etwa 14 %) als im Nüchternzustand festgestellt. Ebenso nimmt auch die interindividuelle Variabilität bei der Ganciclovir-Exposition ab, wenn Valganciclovir zusammen mit einer Mahlzeit eingenommen wird. Valganciclovir wurde in klinischen Studien nur zusammen mit den Mahlzeiten angewendet. Daher wird empfohlen, dass Valganciclovir mit den Mahlzeiten eingenommen wird (siehe Abschnitt 4.2).
Verteilung:
Wegen der raschen Umwandlung von Valganciclovir zu Ganciclovir wurde die Proteinbindung von Valganciclovir nicht bestimmt. Bei Konzentrationen von 0,5 und 51 gg/ml betrug die Plasmaproteinbindung von Ganciclovir 1-2 %. Das Verteilungsvolumen von Ganciclovir im Steady State nach intravenöser Gabe lag bei 0,680 ± 0,161 l/kg (n = 114).
Biotransformation
Valganciclovir wird schnell und umfassend zu Ganciclovir metabolisiert; es wurden keine anderen Metaboliten nachgewiesen. Nach oraler Anwendung von radioaktiv markiertem Ganciclovir (Einmaldosis von 1000 mg) machte kein Metabolit mehr als 1-2 % der in den Fäzes oder im Urin wiedergefundenen Radioaktivität aus.
Elimination
Nach Gabe von Valganciclovir stellt, genau wie nach Gabe von Ganciclovir, die renale Ausscheidung über glomeruläre Filtration und aktive tubuläre Sekretion den Haupteliminationsweg von Valganciclovir dar. Die renale Clearance hat einen Anteil von 81,5 % ± 22 % (n = 70) an der systemischen Clearance von Ganciclovir. Die Halbwertszeit von Ganciclovir aus Valganciclovir beträgt bei HIV- und CMV-seropositiven Patienten 4,1 ± 0,9 Stunden.
Pharmakokinetik in besonderen klinischen Situationen
Patienten mit eingeschränkter Nierenfunktion
Eine Abnahme der Nierenfunktion führte zu einer Abnahme der Clearance von Ganciclovir aus Valganciclovir mit einer entsprechenden Zunahme der terminalen Halbwertszeit. Deshalb ist bei Patienten mit eingeschränkter Nierenfunktion eine Dosisanpassung erforderlich (siehe Abschnitte 4.2 und 4.4).
Dialysepflichtige Patienten
Für dialysepflichtige Patienten kann keine Dosierungsempfehlung für Valganciclovir 450 mg Filmtabletten ausgesprochen werden, weil die für diese Patienten erforderliche individuelle Valganciclovir-Dosis unter der Dosisstärke der 450 mg Tabletten liegt. Daher soll Valganciclovir bei diesen Patienten nicht angewendet werden (siehe Abschnitte 4.2 und 4.4).
Patienten mit eingeschränkter Leberfunktion
Es liegen keine Untersuchungen zur Sicherheit und Wirksamkeit von Valganciclovir Tabletten bei Patienten mit Leberfunktionsstörungen vor. Von einer Beeinflussung der Pharmakokinetik von Ganciclovir durch eine Leberfunktionsstörung ist jedoch eher nicht auszugehen, da die Substanz über die Nieren ausgeschieden wird; demzufolge wird auch keine spezifische Dosierungsempfehlung gegeben.
Kinder und Jugendliche
In einer Phase-II-Studie zur Untersuchung der Pharmakokinetik und Sicherheit bei pädiatrischen Empfängern eines Organtransplantats (im Alter von 4 Monaten bis 16 Jahren, n = 63) wurde Valganciclovir einmal täglich über bis zu 100 Tage verabreicht. Die pharmakokinetischen Parameter waren in Bezug auf die verschiedenen Organe und Altersbereiche ähnlich und mit denen Erwachsener vergleichbar. Populationspharmakokinetische Modellberechnungen ergaben eine Bioverfügbarkeit von ca. 60 %. Die Clearance wurde sowohl von der Körperoberfläche als auch von der Nierenfunktion positiv beeinflusst. Die mittlere totale Clearance lag für einen Patienten mit einer Kreatininclearance von 70,4 ml/min bei 5,3 l/h (88,3 ml/min). Die folgende Tabelle zeigt die mittleren Cmax-, t/2- und AUC-Werte mit der jeweiligen Standardabweichung für die relevanten pädiatrischen Altersgruppen im Vergleich zu Daten von Erwachsenen:
|
PK-Parameter |
Erwachsene* |
Kinder und Jugendliche | ||
|
> 18 Jahre (n = 160) |
< 2 Jahre (n = 17) |
> 2 - < 12 Jahre (n = 21) |
> 12 Jahre (n = 25) | |
|
AUC0-24 h (pg x h/ml) |
46,3 ± 15,2 |
64,3 ± 29,2 |
59,2 ± 15,1 |
50,3 ± 15,0 |
|
Cmax (pg/ml) |
5,3 ± 1,5 |
10,3 ± 3,3 |
9,4 ± 2,7 |
8,0 ± 2,4 |
|
Clearance (l/h) |
12,7 ± 4,5 |
2,5 ± 2,4 |
4,5 ± 2,9 |
6,4 ± 2,9 |
|
t1/2 (h) |
6,5 ± 1,4 |
3,1 ± 1,4 |
4,1 ± 1,3 |
5,5 ± 1,1 |
* Aus dem Studienreport PV 16000 entnommen.
Die einmalige tägliche Valganciclovir-Dosis basierte auf der Körperoberfläche (KO) sowie der Kreatininclearance (CrCl), die mit einer modifizierten Schwartz-Formel berechnet wurde, und errechnete sich anhand der folgenden Formel:
Pädiatrische Dosis (mg) = 7 x KO x CrCl (Berechnung nach der modifizierten Schwartz-Formel) wobei
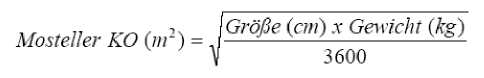
k x Größe (an) Serumkreatinin (mg / dl)
Schwärtz Kreatininclearance (ml / min / 1,73m2)
mit k = 0,45 für Patienten im Alter von < 2 Jahren, 0,55 für Jungen von 2 bis < 13 Jahren sowie Mädchen von 2 bis 16 Jahren und 0,7 für Jungen von 13 bis 16 Jahren.
Die Dosis sollte die Erwachsenendosis von 900 mg nicht überschreiten. Zusätzlich sollte bei einer berechneten Schwartz Kreatininclearance von mehr als 150 ml/min/1,73 m2 der maximale Wert von 150 ml/min/1,73 m2 für die Berechnung verwendet werden. Es wird darauf hingewiesen, dass der Algorithmus zur Berechnung der pädiatrischen Dosis nur auf der Basis von pharmakokinetischen Daten entwickelt wurde und nicht in Wirksamkeits- und Sicherheitsstudien verifiziert wurde (siehe Abschnitt 5.1).
Die Pharmakokinetik von Ganciclovir wurde ebenfalls an 24 Neugeborenen im Alter von 8 bis 34 Tagen mit symptomatischer, kongenitaler CMV-Erkrankung untersucht. Alle Patienten erhielten zweimal täglich intravenös 6 mg/kg Ganciclovir. Anschließend wurden die Patienten zweimal täglich mit oral verabreichtem Valganciclovir behandelt, wobei die Dosis von Valganciclovir Pulver zur Herstellung einer Lösung zum Einnehmen im Bereich von 14 mg/kg bis 20 mg/kg lag. Eine Dosis von zweimal täglich 16 mg/kg Valganciclovir Pulver zur Herstellung einer Lösung zum Einnehmen führte zu einer vergleichbaren Ganciclovir-Exposition wie eine intravenöse Behandlung mit zweimal täglich 6 mg/kg Ganciclovir bei Neugeborenen und auch zu einer ähnlichen Ganciclovir-Exposition wie nach intravenöser Gabe der wirksamen Erwachsenendosis von 5 mg/kg. Die folgende Tabelle zeigt die mittleren AUC-, Cmax- und t/2-Werte mit der jeweiligen Standardabweichung im Vergleich zu Daten von Erwachsenen:
|
PK-Parameter |
Erwachsene |
Neugeborene | |
|
5 mg/kg GAN Einzeldosis (n = 8) |
6 mg/kg GAN zweimal täglich (n = 19) |
16 mg VAL zweimal täglich (n = 19) | |
|
AUC0-X, h (gg x h/ml) |
25,4 ± 4,32 |
- |
- |
|
AUC12 h (mgx h/l) |
- |
38,25 ± 42,7 |
30,1 ± 15,1 |
|
Cmax (gg/ml) |
9,03 ± 1,26 |
12,9 ± 21,5 |
5,44 ± 4,04 |
|
t1/2 (h) |
3,32 ± 0,47 |
2,52 ± 0,55 |
2,98 ± 1,26 |
GAN = Ganciclovir, i.v.
VAL = Valganciclovir, oral
Die pharmakokinetische Modellberechnung sagte für die kennzeichnenden Werte Clearance (l/h), Verteilungsvolumen (l) sowie Bioverfügbarkeit von Ganciclovir bei Neugeborenen Werte von 0,146 x Gewicht1,68, 1,15 x Gewicht bzw. 54 % voraus. Diese Daten sind zu eingeschränkt, um daraus Schlüsse über die Wirksamkeit oder Dosierungsempfehlungen für pädiatrische Patienten mit kongenitaler CMV-Infektion ableiten zu können.
5.3 Präklinische Daten zur Sicherheit
Valganciclovir ist ein Prodrug von Ganciclovir, d. h., mit Ganciclovir beobachtete Wirkungen gelten genauso auch für Valganciclovir. Die in präklinischen Sicherheitsstudien beobachtete Toxizität von Valganciclovir entsprach derjenigen von Ganciclovir und wurde durch Ganciclovir-Expositionen, die der beim Menschen angewendeten Initialdosis entsprachen bzw. darunter lagen, ausgelöst.
Bei diesen toxischen Effekten handelte es sich um irreversible Gonadotoxizität (Hodenzellverlust) und Nephrotoxizität (Urämie, Zelldegeneration) sowie um reversible Myelotoxizität (Anämie, Neutropenie, Lymphozytopenie) und gastrointestinale Toxizität (Nekrose der Schleimhautzellen).
In weiteren Studien hat sich Ganciclovir in vitro im Maus-Lymphom- und Maus-Mikrokern-Test als mutagen erwiesen. Ganciclovir führte zu einer höheren Inzidenz von Tumoren der Vorhautdrüse bei männlichen Mäusen, des Vormagens bei männlichen und weiblichen Mäusen sowie der reproduktiven Gewebe und der Leber bei weiblichen Mäusen.
Darüber hinaus erhöhte die Gabe von Ganciclovir bei Mäusen und Kaninchen die Inzidenz von fetalen Resorptionen und Missbildungen (z. B. Gaumenspalte, Anophtalmie/Mikrophthalmie und Hydrocephalus).
17
Ganciclovir führte auch nachweislich zu einer Beeinträchtigung der Fertilität bei Mäusen und zu einer Hypospermatogenese bei Hunden. Diese karzinogenen und reproduktionstoxischen Wirkungen traten bei systemischem Expositionen auf, die unter oder nur leicht über der bei der klinischen Anwendung zu erwartenden Exposition lagen.
6. PHARMAZEUTISCHE ANGABEN
6.1 Liste der sonstigen Bestandteile
Tablettenkern Mikrokristalline Cellulose Mannitol
Magnesiumstearat (Ph.Eur.) [pflanzlich]
Hochdisperses Siliciumdioxid Crospovidon, Typ A
Filmüberzug der Tablette Opadry II 32K54870 pink enthält:
Hypromellose
Titandioxid
Lactose-Monohydrat
Triacetin
Eisen(III)-oxid
6.2 Inkompatibilitäten
Nicht zutreffend.
6.3 Dauer der Haltbarkeit
2 Jahre
6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Für dieses Arzneimittel sind keine besonderen Lagerungsbedingungen erforderlich.
6.5 Art und Inhalt des Behältnisses PVC/ACLAR/PVC//Aluminium-Blisterpackungen.
Packungsgrößen: 10, 30 und 60 Tabletten
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
7. INHABER DER ZULASSUNG
ratiopharm GmbH
Graf-Arco-Str. 3 89079 Ulm
8. ZULASSUNGSNUMMER(N)
89315.00.00
9. DATUM DER ERTEILUNG DER ZULASSUNG/VERLÄNGERUNG DER ZULASSUNG
Datum der Erteilung der Zulassung: 4. Juli 2014
10. STAND DER INFORMATION
April 2015
11. VERKAUFSABGRENZUNG
Verschreibungspflichtig
19