Wilate 500
ZUSAMMENFASSUNG DER MERKMALE DES ARZNEIMITTELS
1. BEZEICHNUNG DES ARZNEIMITTELS
Wilate 500, 500 I.E. VWF/500 I.E. FVIII, Pulver und Lösungsmittel zur Herstellung einer intravenösen Injektionslösung.
Wilate 1000, 1000 I.E. VWF/1000 I.E. FVIII, Pulver und Lösungsmittel zur Herstellung einer intravenösen Injektionslösung.
2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
Wilate, hergestellt aus menschlichem Plasma, ist ein Pulver und Lösungsmittel zur Herstellung einer intravenösen Injektionslösung, welche nominal 500 I.E./1000 I.E. humanen von Willebrand-Faktor (VWF) und humanen Blutgerinnungsfaktor VIII (FVIII) pro Flasche enthält.
Wilate enthält etwa 100 I.E./ml humanen VWF nach Auflösen in 5 ml/ 10 ml Wasser für Injektionszwecke mit 0,1% Polysorbat 80.
Die spezifische Aktivität von Wilate beträgt mehr als 67 I.E. VWF:RCo/mg Protein.
Die Bestimmung der VWF-Aktivität (I.E.) erfolgt als Bestimmung der Ristocetin-Kofaktor-Aktivität (VWF:RCo) unter Verwendung des Internationalen Standards für VWF-Konzentrat (WHO).
Wilate enthält etwa 100 I.E./ml humanen FVIII nach Auflösen in 5 ml/ 10 ml Wasser für Injektionszwecke mit 0,1% Polysorbat 80.
Die Bestimmung der FVIII-Aktivität (I.E.) erfolgt mittels chromogenem Testverfahren gem. Europäischem Arzneibuch. Die spezifische Aktivität von Wilate beträgt mehr als 67 I.E. FVIII:C/mg Protein.
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
3. DARREICHUNGSFORM
Pulver und Lösungsmittel zur Herstellung einer intravenösen Injektionslösung. Pulver, gefriergetrocknet: weißes oder leicht gelbliches Pulver oder krümelige Substanz.
4. KLINISCHE ANGABEN
4.1 Anwendungsgebiete
Von Willebrand-Syndrom
Vorbeugung und Behandlung von Blutungen oder Behandlung von Blutungen bei chirurgischen Eingriffen bei Patienten mit von Willebrand-Syndrom (VWS), wenn die Behandlung mit Desmopressin (DDAVP) allein unwirksam oder kontraindiziert ist.
Hämophilie A
Therapie und Prophylaxe von Blutungen bei Patienten mit Hämophilie A (angeborener FVIII-Mangel).
4.2 Dosierung und Art der Anwendung
Die Behandlung sollte nur unter Aufsicht eines in der Behandlung von Blutgerinnungsstörungen erfahrenen Arztes durchgeführt werden. Die Arzneimittel sind zum einmaligen Gebrauch bestimmt und der gesamte Inhalt sollte verwendet werden. Nicht verwendete Arzneimittel sind entsprechend den nationalen Anforderungen zu entsorgen.
Von Willebrand-Syndrom
Das Verhältnis von VWF-Ristocetin-Kofaktor-Aktivität zu FVIII-Aktivität ist 1:1. Generell erhöht eine Einheit VWF:RCo und FVIII:C/kg KG die Aktivität um 1,5-2 I.E./dl des jeweiligen Proteins. Normalerweise werden ca. 20-50 I.E. Wilate/kg KG gegeben, um eine ausreichende Hämostase zu erreichen. Dies erhöht den VWF:RCo und FVIII:C im Patienten um ca. 30-100%.
Als Initialdosis können 50-80 I.E. Wilate/kg KG nötig sein, insbesondere bei Patienten mit von Willebrand-Syndrom Typ 3, bei denen zur Erhaltung eines ausreichenden Plasmaspiegels höhere Dosierungen nötig sein können als bei anderen Typen des von Willebrand-Syndroms.
Kinder und Jugendliche
Zur Anwendung von Wilate bei Kindern unter 6 Jahren liegen keine ausreichenden Daten vor.
Prävention von Blutungen bei Operationen und schweren Traumata:
Zur Prävention von Blutungen im Rahmen von Operationen sollte die Gabe von Wilate 1-2 Stunden vor dem Eingriff erfolgen.
Es sollten VWF:RCo-Plasmaspiegel von > 60 I.E./dl (> 60 %) und FVIII:C-Plasmaspiegel von > 40 I.E./dl (> 40 %) erreicht werden.
Während der Behandlung sollte eine entsprechende Dosis alle 12-24 Stunden gegeben werden. Die Dosierung und die Dauer der Therapie richten sich nach der klinischen Wirksamkeit, der Art und der Schwere der Blutung, sowie nach den VWF:RCo- und FVIII:C- Plasmaspiegeln.
Bei Verwendung eines FVIII-haltigen VWF-Präparates sollte der behandelnde Arzt berücksichtigen, dass Patienten mit von Willebrand-Syndrom, die über einen längeren Zeitraum mit FVIII-haltigen VWF-Produkten behandelt werden, deutlich erhöhte FVIII:C-Plasmaspiegel aufweisen können. Diese können das Risiko einer Thrombose erhöhen, insbesondere bei Patienten mit bekannten klinischen und labordiagnostischen Risikofaktoren. Im Falle sehr hoher FVIII:C-Plasmaspiegel sollte eine Dosisreduzierung und/oder Verlängerung des Dosierungsintervalls bzw. der Einsatz von VWF-Präparaten mit niedrigem FVIII-Gehalt erwogen werden.
Vorbeugende Dauerbehandlung (Prophylaxe):
Für eine Langzeitprophylaxe bei VWS-Patienten sind 20 - 40 I.E. Wilate pro kg Körpergewicht zwei- oder dreimal wöchentlich zu verabreichen. In manchen Fällen, zum Beispiel bei Patienten mit gastrointestinalen Blutungen, können höhere Dosen erforderlich sein.
Hämophilie A
Dosis und Dauer der Therapie hängen vom Schweregrad des FVIII-Mangels sowie vom Ort und Ausmaß der Blutung und dem klinischen Zustand des Patienten ab.
Die Anzahl der zu verabreichenden FVIII-Einheiten ist in Internationalen Einheiten angegeben (I.E.), für die der aktuelle WHO-Standard für FVIII gilt. FVIII-Aktivität im Plasma wird entweder in Prozentsatz (bezogen auf normales Humanplasma) oder in Internationalen Einheiten (bezogen auf einen Internationalen Standard für FVIII im Plasma) angegeben. Eine Internationale Einheit (I.E.) des FVIII vom Menschen entspricht der FVIII-Menge in 1 ml normalem Humanplasma.
Bedarfsbehandlung:
Die Dosisberechnung basiert auf dem empirischen Befund, dass 1 I.E. FVIII vom Menschen pro kg KG die FVIII-Aktivität im Mittel um 1,5% bis 2% der normalen Aktivität erhöht.
Die notwendige Dosis kann mit folgender Formel bestimmt werden:
Erforderliche Einheiten = KG (kg) x gewünschter FVIII-Anstieg (%) (I.E./dl) x 0,5 I.E./kg
Die zu verabreichende Menge und die Häufigkeit der FVIII-Gabe sollten sich im Einzelfall immer an der klinischen Wirksamkeit orientieren.
Im Falle der aufgeführten hämorrhagischen Ereignisse sollte die FVIII-Aktivität in dem entsprechenden Zeitraum nicht unter den angegebenen Plasmaspiegel (in % von normal oder IE/dl) sinken. Die nachfolgende Tabelle kann als Richtlinie zur Festlegung der Dosis bei Blutungsepisoden und chirurgischen Eingriffen dienen.
|
Blutungsgrad / Art der chirurgischen Intervention |
Notwendiger FVIII-Spiegel (%) (I.E./dl) |
Behandlungshäufigkeit (Stunden) und -dauer (Tage) |
|
Blutungen Frühstadium von Gelenk-und Muskelblutungen, oder Blutungen im Mund |
20 - 40 |
Infusion alle 12 - 24 Stunden wiederholen. Für mindestens 1 Tag bzw. bis die durch Schmerzen angezeigte Blutung sistiert oder eine Heilung erreicht ist. |
|
Größere Blutungen, Gelenkblutungen, Muskelblutungen oder Hämatome |
30-60 |
Infusion alle 12-24 Stunden wiederholen. Für 3 - 4 Tage oder länger bis Schmerzen und gegebenenfalls Bewegungseinschränkungen beseitigt sind. |
|
Lebensbedrohliche Blutungen |
60-100 |
Infusion alle 8-24 Stunden wiederholen bis die Bedrohung abgewendet ist. |
|
Operationen Kleine Eingriffe (einschließlich Zahnextraktionen) |
30 - 60 |
Alle 24 Stunden, für mindestens 1 Tag bzw. bis Heilung erfolgt. |
|
Große Operationen |
80-100 (prä- und postoperativ) |
Infusion alle 8-24 Stunden wiederholen bis adäquate Wundheilung erfolgt. Anschließend noch weitere 7 Tage therapieren, um einen FVIII-Spiegel von 30%-60% (I.E./dl) aufrecht zu erhalten. |
Vorbeugende Dauerbehandlung (Prophylaxe):
Ist eine blutungsvorbeugende Dauerbehandlung angezeigt, werden bei schwerer Hämophilie A 20 - 40 I.E. Wilate pro kg KG zwei- bis dreimal wöchentlich verabreicht. Auch hier ist eine individuelle Anpassung der Dosierung je nach klinischer Situation erforderlich. In manchen Fällen, besonders bei jüngeren Patienten, können kürzere Dosierungszeiträume oder höhere Dosen erforderlich sein.
Kontinuierliche Infusion:
Vor Operationen sollte eine pharmakokinetische Analyse durchgeführt werden, um die Clearance abschätzen zu können. Die initiale Infusionsrate kann folgendermaßen errechnet werden:
Infusionsrate (I.E./kg/h) = Clearance (ml/kg/h) x gewünschter/gemessener Spiegel (I.E./ml)
Nach den ersten 24 Stunden der kontinuierlichen Infusion sollte die Clearance täglich unter Verwendung der o.g. Gleichung mit dem gemessenen Wert und der verwendeten Infusionsrate errechnet werden.
Während der Behandlung sollte der FVIII-Spiegel entsprechend bestimmt werden, um die zu verabreichende Dosis und die Häufigkeit wiederholter Infusionen zu steuern. Insbesondere bei größeren chirurgischen Eingriffen ist eine genaue Überwachung der Substitutionstherapie (FVIII-Aktivität im Plasma) unerlässlich. Einzelne Patienten können in ihrem Ansprechen auf FVIII variieren, wobei unterschiedliche Werte der in-vivo Recovery und Halbwertzeit erreicht werden.
Zuvor unbehandelte Patienten
Zurzeit vorliegende Daten werden in Abschnitt 4.8 beschrieben.
Kinder und Jugendliche
Zur Anwendung von Wilate bei Hämophilie A bei Kindern unter 6 Jahren liegen keine ausreichenden Daten vor.
Art der Anwendung
Intravenöse Anwendung
Die Injektions- bzw. Infusionsrate sollte 2-3 ml pro Minute nicht überschreiten. Hinweise zur Rekonstitution des Arzneimittels vor der Anwendung siehe Abschnitt 6.6.
4.3 Gegenanzeigen
Überempfindlichkeit gegen die Wirkstoffe oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Wie bei allen intravenös zu verabreichenden Plasmaproteinprodukten ist eine Überempfindlichkeit (allergische Reaktionen) möglich. Patienten sollten daher während der gesamten Injektionszeit überwacht werden.
Patienten sollten über die Zeichen von Überempfindlichkeit oder allergische Reaktionen (wie z.B. Ausschlag, generalisierte Urtikaria, Engegefühl in der Brust, Stridor, Hypotonie bis hin zum anaphylaktischen Schock) aufgeklärt werden.
Beim Auftreten allergischer Symptome sollen Patienten die Behandlung sofort abbrechen und einen Arzt aufsuchen. Im Falle eines Schocks sind die aktuellen medizinischen Richtlinien zur Schocktherapie anzuwenden.
Zur Prävention von Infektionen, die durch die Verwendung von Arzneimitteln aus humanem Blut oder Plasma übertragen werden können, werden Standardmaßnahmen ergriffen. Diese Maßnahmen beinhalten Auswahl der Spender, Testung der Einzelspenden und des Plasmapools auf spezifische Infektionsmarker und effektive Verfahrensschritte zur Inaktivierung/Entfernung von Viren.
Trotzdem kann die Möglichkeit der Übertragung infektiöser Agenzien bei der Anwendung von Arzneimitteln, die aus menschlichem Blut oder Plasma hergestellt wurden, nicht völlig ausgeschlossen werden. Dies gilt auch für unbekannte oder neu auftretende Viren oder andere Infektionserreger.
Die Maßnahmen werden als wirksam für umhüllte Viren wie Humanes Immundefizienz-Virus (HIV), Hepatitis-B-Virus (HBV) und Hepatitis-C-Virus (HCV) und für das nicht umhüllte Hepatitis-A-Virus angesehen.
Die Maßnahmen können von begrenzter Wirksamkeit gegen nicht umhüllte Viren wie Parvovirus B19 sein.
Eine Parvovirus B19-Infektion kann für schwangere Frauen (Infektion des Fetus) und Patienten mit Immunschwäche oder mit gesteigerter Produktion von roten Blutkörperchen (z.B. bei hämolytischer Anämie) schwerwiegende Folgen haben.
Bei jeder Verabreichung von Wilate müssen der Name und die Chargennummer des Präparates dokumentiert werden, um die Chargennachverfolgung zum Patienten zu gewährleisten.
Für Patienten, die regelmäßig Präparate aus menschlichem Blut oder Plasma (z.B. VWF/FVIII-haltige Präparate) erhalten, wird ein angemessener Hepatitisimpfschutz (Hepatitis A und B) empfohlen.
Von Willebrand-Syndrom
Bei Verwendung eines FVIII-haltigen VWF-Präparates sollte der behandelnde Arzt berücksichtigen, dass eine über längere Zeit fortgesetzte Therapie zu einem übermäßig hohen Anstieg von FVIII:C führen kann. Bei Patienten, die FVIII-haltige VWF-Präparate erhalten, sollte daher der FVI11:C - Plasmaspiegel überwacht werden, um anhaltend übermäßig hohe Spiegel zu vermeiden, die mit einem erhöhten Thromboserisiko einhergehen können.
Das Auftreten von thrombotischen Komplikationen, insbesondere bei Patienten mit bekannten klinischen oder labordiagnostisch nachgewiesenen Risikofaktoren, ist für FVIII-haltige VWF-Präparate beschrieben. Daher sind Patienten mit Risikofaktoren auf frühe Zeichen einer Thrombose zu überwachen. Generell sollte eine Thrombosephrophylaxe entsprechend den aktuellen Empfehlungen durchgeführt werden.
Patienten mit von Willebrand-Syndrom, speziell Typ 3, können neutralisierende Antikörper (Inhibitoren) gegen VWF bilden. Falls der erwartete Anstieg des VWF:RCo-Plasmaspiegels nicht erreicht wird, oder falls die Blutung mit einer entsprechenden Dosis nicht kontrolliert werden kann, sollte ein Test auf VWF-Antikörper durchgeführt werden. Bei Patienten mit hochtitrigem Inhibitor kann die VWF-Therapie ineffektiv sein, und andere therapeutische Möglichkeiten sollten in Betracht gezogen werden. Die Behandlung solcher Patienten sollte durch Ärzte erfolgen, die Erfahrung in der Behandlung von Blutgerinnungsstörungen haben.
Hämophilie A
Überempfindlichkeit
Allergische Überempfindlichkeitsreaktionen sind mit Wilate möglich. Das Präparat enthält zusätzlich zu Faktor VIII Spuren anderer Humanproteine. Wenn Überempfindlichkeitssymptome auftreten, sollten die Patienten angewiesen werden, das Arzneimittel sofort abzusetzen und ihren Arzt zu kontaktieren. Patienten sollten über Frühzeichen von Überempfindlichkeitsreaktionen aufgeklärt werden, wie zum Beispiel Nesselausschlag, generalisierte Urtikaria, Engegefühl in der Brust, Keuchen (Atemnot) und Blutdruckabfall bis hin zum anaphylaktischen Schock.
Im Fall eines Schocks soll eine Schocktherapie nach aktuellem medizinischen Standard durchgeführt werden.
Inhibitoren
Die Bildung von neutralisierenden Antikörpern (Inhibitoren) gegen FVIII ist eine bekannte Komplikation bei der Behandlung von Hämophilie-A-Patienten. Diese Inhibitoren sind normalerweise IgG-Immunglobuline, die gegen die prokoagulative FVIII-Aktivität gerichtet sind und werden in modifizierten Bethesda-Einheiten (BE) pro ml Plasma quantifiziert. Das Risiko der Inhibitorbildung steht im Zusammenhang mit der Menge des applizierten FVIII, wobei das größte Risiko innerhalb der ersten 20 Behandlungstage besteht. In seltenen Fällen treten Inhibitoren auch nach den ersten 100 Behandlungstagen auf.
Bei Patienten mit Inhibitoren in der Anamnese und mehr als 100 Expositionstagen wurden bei Umstellung auf andere FVIII-Präparate Fälle von wiederkehrenden, niedrigtitrigen Inhibitoren beobachtet. Daher wird empfohlen, alle Patienten nach einer Umstellung sorgfältig auf die Entstehung von Inhibitoren zu überwachen.
Im Allgemeinen sollten Patienten, die mit Blutgerinnungsfaktor VIII behandelt werden, durch angemessene klinische Beobachtungen und Labortests sorgfältig auf die Entwicklung eines Inhibitors hin überwacht werden. Wenn die erwarteten Faktor VIII-Spiegel im Plasma nicht erzielt werden, oder die Blutung nicht durch eine angemessene Dosis unter Kontrolle gebracht werden kann, sollte auf das Vorhandensein von Faktor VIII-Inhibitoren getestet werden. Bei Patienten mit einem hohen Inhibitorspiegel ist eine Faktor VIII-Therapie möglicherweise nicht wirksam und es sollten andere Behandlungsoptionen in Betracht gezogen werden. Die Behandlung dieser Patienten sollte von Ärzten geleitet werden, die im Umgang mit Hämophilie und Faktor VIII-Inhibitoren Erfahrung haben.
Wilate 500 (500 I.E. VWF und FVIII) enthält pro Durchstechflasche bis zu 2,55 mmol (58,7 mg) Natrium. Wilate 1000 (1000 I.E. VWF und FVIII) enthält pro Durchstechflasche bis zu 5,1 mmol (117,3 mg) Natrium. Dies ist bei Patienten zu berücksichtigen, die auf eine natriumarme Ernährung achten müssen.
4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Wechselwirkungen mit anderen Arzneimitteln sind nicht bekannt.
4.6 Fertilität, Schwangerschaft und Stillzeit
Reproduktionsstudien an Tieren wurden mit VWF/FVIII nicht durchgeführt.
Von Willebrand-Syndrom
Gesicherte Daten zur Anwendung bei schwangeren oder stillenden Frauen liegen nicht vor. Wilate sollte bei schwangeren oder stillenden Frauen mit von WillebrandSyndrom nur angewendet werden, wenn dies unbedingt erforderlich ist. Es sollte berücksichtigt werden, dass der Geburtsvorgang ein erhöhtes Blutungsrisiko bei Frauen mit von Willebrand-Syndrom darstellt.
Hämophilie A
Aufgrund des seltenen Auftretens von Hämophilie A bei Frauen liegen keine gesicherten Daten über die Anwendung während der Schwangerschaft und Stillzeit vor. Daher sollte Wilate bei schwangeren oder stillenden Frauen nur angewendet werden, wenn dies unbedingt erforderlich ist.
4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Wilate hat keinen Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
4.8 Nebenwirkungen
Zusammenfassung des Sicherheitsprofils
Überempfindlichkeit oder allergische Reaktionen (wie z.B. Angioödem, Brennen und Stechen an der Injektionsstelle, Schüttelfrost, Hitzegefühl, generalisierte Urtikaria, Kopfschmerzen, Ausschlag, Hypotonie, Lethargie, Übelkeit, nervöse Unruhe, Tachykardie, Engegefühl in der Brust, Zittern, Erbrechen, Stridor) wurden selten bei Patienten beobachtet, in Einzelfällen bis zur Ausbildung eines anaphylaktischen Schocks.
In seltenen Fällen wurde eine erhöhte Körpertemperatur festgestellt.
Von Willebrand-Syndrom
Bei Patienten mit von Willebrand-Syndrom, speziell Typ 3 - Patienten, können in sehr seltenen Fällen Hemmkörper gegen den VWF gebildet werden. Das Auftreten solcher Inhibitoren manifestiert sich in einer unzureichenden klinischen Wirksamkeit. Diese Inhibitoren können in enger Assoziation mit anaphylaktischen Reaktionen auftreten. Deswegen sollten Patienten mit anaphylaktischen Reaktionen auf Hemmkörper getestet werden.
Es wird empfohlen, in diesen Fällen ein spezialisiertes Hämophiliezentrum aufzusuchen.
Bislang wurden keine Fälle von VWF-Inhibitoren in klinischen Studien oder in der Anwendung nach der Zulassung von Wilate berichtet.
Es besteht ein Risiko für thrombotische Ereignisse, besonders bei Patienten mit bekannten klinischen oder labordiagnostisch nachgewiesenen Risikofaktoren.
Eine Thromboseprophylaxe sollte entsprechend den aktuellen Empfehlungen eingeleitet werden.
Bei Patienten mit von Willebrand-Syndrom, die FVIII-haltige VWF-Produkte erhalten, können anhaltend hohe FVIII:C-Plamaspiegel zu einem erhöhten Thromboserisiko führen.
Hämophilie A
Bei Hämophilie-A-Patienten können sich in seltenen Fällen Hemmkörper gegen FVIII bilden. Das Auftreten solcher Inhibitoren manifestiert sich in einer unzureichenden klinischen Wirksamkeit. Es wird empfohlen, in diesen Fällen ein spezialisiertes Hämophiliezentrum aufzusuchen.
Die Erfahrung mit Wilate bei nicht behandelten Patienten ist begrenzt. In einer klinischen Studie mit 24 unbehandelten Patienten mit mindestens 50 Expositionstagen wurden bei nur drei Patienten nach der Behandlung mit Wilate persistierende und klinisch relevante Inhibitoren mit über 5 Bethesda-Einheiten (BE) festgestellt. Drei Patienten entwickelten niedrigtitrige und transiente Inhibitoren ohne klinische Auswirkungen. Bei zwei Patienten wurden niedrigtitrige Inhibitoren nur bei einer einzigen Messung festgestellt, die bei wiederholter Messung nicht bestätigt wurden.
Siehe auch 4.2. Es wurden keine Inhibitor Entwicklungen bei vorher behandelten Patienten beobachtet.
Zu Informationen zur Virussicherheit siehe Abschnitt 4.4. Tabellarische Auflistung von Nebenwirkungen
Die nachfolgende Tabelle entspricht der MedDRA-Systemorganklassifizierung (SOC und bevorzugter Begriff).
Die Häufigkeiten wurden gemäß folgender Konvention bestimmt: sehr häufig (>1/10); häufig (>1/100 bis <1/10); gelegentlich (>1/1.000 bis <1/100); selten (>1/10.000 bis <1/1.000); sehr selten (<1/10.000), nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
|
Organklasse |
Gelegentlich |
Selten |
Sehr selten |
|
Erkrankungen des Immunsystems |
Überempfindlichkeits reaktion |
Anaphylaktischer Schock | |
|
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort |
Fieber | ||
|
Untersuchungen |
FVIII-Inhibitoren |
VWF- Inhibitoren |
Beschreibung einzelner Nebenwirkungen
Beschreibung einzelner Nebenwirkungen siehe Abschnitt 4.4 Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-RisikoVerhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel, Paul-Ehrlich-Institut, Paul-EhrlichStraße 51-59, 63225 Langen, Telefon: +49 6103 77 0, Telefax: +49 6103 77 1234, Website: www.pei.de anzuzeigen.
4.9 Überdosierung
Symptome von Überdosierung mit humanem VWF oder FVIII wurden bislang nicht beobachtet. Thromboembolische Komplikationen können bei massiver Überdosierung auftreten.
5. PHARMAKOLOGISCHE EIGENSCHAFTEN
5.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Antihaemorrhagika: Blutgerinnungsfaktoren:
Von Willebrand-Faktor human und Faktor VIII human in Kombination ATC-Code: B02BD06
Von Willebrand-Syndrom
VWF (im Konzentrat) ist ein normaler Bestandteil des menschlichen Plasmas und verhält sich wie körpereigener VWF.
Die Verabreichung von VWF erlaubt die Korrektur von Blutgerinnungsstörungen bei Patienten, die an VWF-Mangel leiden, auf zwei Ebenen:
- VWF stellt die Plättchen-Adhäsion an das vaskuläre Sub-Endothelium an der Stelle der vaskulären Verletzung wieder her (da es an das vaskuläre Sub-Endothelium und an die Plättchenmembran bindet). VWF sorgt für die primäre Hämostase, was durch die Verkürzung der Blutungszeit sichtbar wird. Dieser Effekt tritt unmittelbar auf und hängt zum Großteil vom Grad der Polymerisation des Proteins ab;
- VWF führt zur verzögerten Korrektur eines assoziierten FVIII-Mangels. Intravenös verabreichter VWF bindet an den endogenen FVIII (der normalerweise vom Patienten produziert wird) und stabilisiert diesen, indem er eine rasche Degradation verhindert.
Die Gabe von reinem VWF (VWF mit geringem FVIII-Gehalt) stellt den FVIII:C-Plasmaspiegel nach der ersten Infusion erst mit Verzögerung wieder her.
Die Gabe von FVIII:C-haltigen VWF-Präparaten stellt den FVIII: C-Plasmaspiegel nach der ersten Infusion sofort wieder her.
Neben seiner Schutzfunktion für den FVIII vermittelt der VWF die Adhäsion der Plättchen an einem Verletzungsort und spielt eine Rolle bei der Aggregation der Plättchen.
Hämophilie A
Der Faktor VIII/von Willebrand-Faktor-Komplex besteht aus zwei Molekülen (FVIII und VWF) mit unterschiedlichen physiologischen Funktionen.
Infundierter FVIII bindet an den VWF im Kreislauf des Patienten.
Aktivierter FVIII ist ein Kofaktor für aktivierten Faktor IX und beschleunigt die Aktivierung von Faktor X zu aktiviertem Faktor X (FXa). Faktor Xa wandelt Prothrombin zu Thrombin, welches dann Fibrinogen zu Fibrin umwandelt, wodurch sich ein Gerinnsel bilden kann. Hämophilie A ist in der Regel eine geschlechtsbezogene erbliche Blutgerinnungsstörung, die auf verringerte Spiegel von FVIII zurückzuführen ist. Sie kann zu massiven Blutungen in Gelenken, Muskeln oder inneren Organen führen, die entweder spontan auftreten oder als Folge von Verletzungen oder chirurgischen Eingriffen.
Durch die Substitutionstherapie wird der FVIII-Spiegel im Plasma erhöht und somit ist eine vorübergehende Korrektur des Faktormangels und der Blutungstendenz möglich.
5.2 Pharmakokinetische Eigenschaften
Von Willebrand-Syndrom
Der VWF (im Konzentrat) ist ein normaler Bestandteil des menschlichen Plasmas und verhält sich wie der körpereigene VWF.
Basierend auf der Meta-Analyse von drei pharmakokinetischen Studien mit auswertbaren Daten von 24 Patienten wurden die folgenden Ergebnisse beobachtet:
|
Alle VWS-Typen |
VWS-Typ 1 |
VWS-Typ 2 |
VWS-Typ 3 | |||||||||||||||||
|
Parameter |
N |
Mittel |
SD |
Min. |
Max. |
N |
Mittel |
SD |
Min. |
Max. |
N |
Mittel |
SD |
Min. |
Max. |
N |
Mittel |
SD |
Min. |
Max. |
|
Recovery (%/IE/kg) |
24 |
1,56 |
0,48 |
0,90 |
2,93 |
2 |
1,19 |
0,07 |
1,14 |
1,24 |
5 |
1,83 |
0,86 |
0,98 |
2,93 |
17 |
1,52 |
0,32 |
0,90 |
2,24 |
|
AUC (0-inf) (h*%) |
23 |
1981 |
960 |
593 |
4831 |
2 |
2062 |
510 |
1701 |
2423 |
5 |
2971 |
1383 |
1511 |
4831 |
16 |
1662 |
622 |
593 |
2606 |
|
T 1/2 (h) |
24 |
23,3 |
12,6 |
7,4 |
58,4 |
2 |
39,7 |
18,3 |
26,7 |
52,7 |
5 |
34,9 |
16 |
17,5 |
58,4 |
17 |
18 |
6,2 |
7,4 |
30,5 |
|
MRT (h) |
24 |
33,1 |
19 |
10,1 |
89,7 |
2 |
53,6 |
25,9 |
35,3 |
71,9 |
5 |
53,5 |
24,6 |
27,8 |
89,7 |
17 |
24,7 |
8,5 |
10,1 |
37,7 |
|
Clearance (mL/h/kg) |
24 |
3,29 |
1,67 |
0,91 |
7,41 |
2 |
2,66 |
0,85 |
2,06 |
3,27 |
5 |
1,95 |
1,02 |
0,91 |
3,31 |
17 |
3,76 |
1,69 |
1,83 |
7,41 |
SD = Standardabweichung
AUC (area under the curve) = Fläche unter der Konzentrations-ZeitKurve bezogen auf das KG T 1/2 = Halbwertzeit
MRT mean residence time = Mittlere Verweildauer
Hämophilie A
FVIII (im Konzentrat) ist ein normaler Bestandteil des menschlichen Plasmas und verhält sich wie körpereigener FVIII.
Nach Injektion des Produktes bleiben ungefähr 2/3 bis 3/4 des FVIII in Zirkulation. Die FVIII-Aktivität im Plasma sollte zwischen 80%-120% der erwarteten FVIII-Aktivität liegen.
Die FVIII-Aktivität verringert sich exponentiell in zwei Phasen. Zunächst wird der FVIII in den Körperflüssigkeiten verteilt und wird mit einer Halbwertzeit von 3 bis 6 Stunden aus dem Plasma entfernt. In der folgenden langsameren Phase variiert die Halbwertzeit zwischen 8 und 18 Stunden, mit einem Durchschnitt von 15 Stunden. Dies entspricht der biologischen Halbwertzeit.
Für Wilate wurden in einer Pharmakokinetikstudie die folgenden Resultate mit 12 Patienten ermittelt (chromogene Methode, Doppelbestimmung):
|
Parameter |
Initialwerte |
6-Monatswerte | ||
|
Mittelwert |
SD |
Mittelwert |
SD | |
|
Recovery %/IE/kg |
FVIIIiC 2,27 |
1,20 |
FVIIIiC 2,26 |
1,19 |
|
AUCnorm % 1 h/IU/kg |
FVIIIiC 31,3 |
7,31 |
FVIIIiC 33,8 |
10,9 |
|
Halbwertzeit (h) |
FVIII:C 11,2 |
2,85 |
FVIIIiC 11,8 |
3,37 |
|
MRT (h) |
FVIII:C 15,3 |
3,5 |
FVIIIiC 16,3 |
4,6 |
|
Clearance |
FVIII:C |
0,86 |
FVIIIiC |
1,04 |
|
mL/h/kg |
3,37 |
3,24 | ||
SD = Standardabweichung
AUC (area under the curve) = Fläche unter der Konzentrations
Zeit-Kurve bezogen auf das KG
MRT mean residence time = Mittlere Verweildauer
5.3 Präklinische Daten zur Sicherheit
Die in Wilate enthaltenen arzneilich wirksamen Bestandteile VWF und FVIII sind normale Bestandteile des menschlichen Plasmas und verhalten sich wie körpereigene Faktoren.
Herkömmliche Tierversuche mit diesen Faktoren würden keine verwertbaren Informationen über die bestehenden klinischen Erfahrungen hinaus erbringen und sind somit nicht erforderlich.
6. PHARMAZEUTISCHE ANGABEN
6.1 Liste der sonstigen Bestandteile
Pulver: Natriumchlorid, Glycin, Saccharose, Natriumcitrat und Calciumchlorid. Lösungsmittel: Wasser für Injektionszwecke mit 0,1% Polysorbat 80.
6.2 Inkompatibilitäten
Da keine Kompatibilitätsstudien durchgeführt wurden, darf Wilate nicht mit anderen Medikamenten gemischt oder gleichzeitig mit anderen intravenösen Zubereitungen im gleichen Schlauchsystem verabreicht werden.
Zur intravenösen Gabe soll ausschließlich das beigefügte Injektionsset verwendet werden, da eine Adsorption von FVIII/VWF an den Innenoberflächen anderer Injektionssets nicht ausgeschlossen werden kann.
6.3 Dauer der Haltbarkeit
3 Jahre.
Die Haltbarkeit der gebrauchsfertigen Lösung wurde für 4 Stunden bei Raumtemperatur (bis max. 25°C) nachgewiesen. Aus mikrobiologischer Sicht ist die gebrauchsfertige Lösung jedoch umgehend nach Herstellung zu verwenden.
6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Pulver und Lösungsmittel im Kühlschrank (2-8°C) lagern und in der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen. Nicht einfrieren.
Wilate kann auch bei Raumtemperatur (bis max. 25°C) 2 Monate aufbewahrt werden. In diesem Fall läuft die Haltbarkeit des Produktes 2 Monate nach der ersten Entnahme aus dem Kühlschrank ab. Das neue Haltbarkeitsdatum muss vom Patienten außen auf dem Karton vermerkt werden.
Die gebrauchsfertige Lösung ist nur zum einmaligen Gebrauch bestimmt. Nicht verbrauchte Lösung verwerfen.
Aufbewahrungsbedingungen nach Rekonstitution, siehe Abschnitt 6.3.
6.5 Art und Inhalt des Behältnisses
Pulver und Lösungsmittel zur Herstellung einer intravenösen Injektionslösung. Packungsgrößen:
Wilate 500 (500 I.E. VWF und 500 I.E. FVIII)
1 Packung enthält:
1 Durchstechflasche mit Pulver, Typ-I-Glas, verschlossen mit einem
Brombutylstopfen und einer Flip-off-Bördelkappe
1 Durchstechflasche mit Lösungsmittel (5 ml Wasser für Injektionszwecke mit 0,1% Polysorbat 80)
1 Gerätesatz bestehend aus 1 Transferset [Mix2VialTM], 1 Flügelkanüle, 1 Einmalspritze
2 Alkoholtupfer
Wilate 1000 (1000 I.E. VWF und 1000 I.E. FVIII)
1 Packung enthält:
1 Durchstechflasche mit Pulver, Typ-I-Glas, verschlossen mit einem
Brombutylstopfen und einer Flip-off Bördelkappe
1 Durchstechflasche mit Lösungsmittel (10 ml Wasser für Injektionszwecke mit 0,1% Polysorbat 80)
1 Gerätesatz bestehend aus 1 Transferset [Mix2VialTM], 1 Flügelkanüle, 1 Einmalspritze
2 Alkoholtupfer
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung 1
• Die gebrauchsfertige Injektionslösung in der Spritze ist klar oder leicht schillernd (opaleszent). Verwenden Sie keine Lösungen, die trübe aussehen oder Rückstände enthalten.
• Das gebrauchsfertige Präparat unmittelbar nach dem Auflösen verwenden, um mikrobielle Verunreinigungen zu verhindern.
• Verwenden Sie bitte ausschließlich das mitgelieferte Injektionszubehör. Die Anwendung anderer Injektions-/Infusionsbestecke kann mit Risiken verbunden sein oder die Wirksamkeit beeinträchtigen.
Anleitung für das Auflösen:
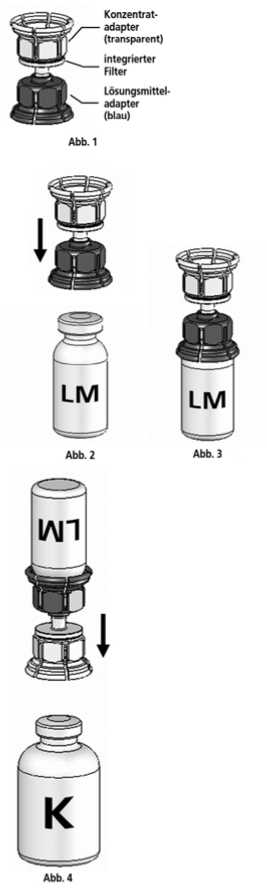
1. Lösungsmittel und Konzentrat in den ungeöffneten Flaschen auf Zimmertemperatur bringen. Nicht direkt aus dem Kühlschrank verwenden.
2. Die Schutzkappen von der Konzentratflasche und Lösungsmittelflasche entfernen und die Gummistopfen beider Flaschen mit einem Alkoholtupfer desinfizieren.
3. Die Lösungsmittelflasche auf eine feste, ebene Fläche stellen. Das in Abb. 1 beschriebene Mix2VialTM-Set mit dem blauen Adapter auf die Lösungsmittelflasche (LM) aufsetzen und bis zum Anschlag nach unten drücken (Abb. 2+3).
4. Die Konzentratflasche (K) auf eine feste, ebene Fläche stellen. Die Lösungsmittelflasche (LM) mit dem Mix2Vial™-Set umdrehen und senkrecht mit dem transparenten Ende auf die Konzentratflasche (K) aufsetzen und bis zum Anschlag nach unten drücken (Abb. 4). Das Vakuum in der Konzentratflasche saugt das Lösungsmittel an.
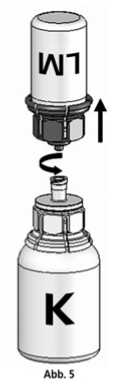
5. Die Konzentratflasche mit Mix2VialTM-Set und der verbundenen Lösungsmittelflasche leicht schwenken (nicht schütteln) bis das Produkt vollständig gelöst ist. Das Konzentrat löst sich bei Zimmertemperatur spätestens nach 10 Minuten vollständig. Dabei ist eine leichte Schaumbildung möglich, die sich auflösen wird. Die Lösungsmittelflasche zusammen mit dem blauen Adapter von der Konzentratflasche abdrehen (Abb. 5) und die Lösungsmittelflasche mit dem blauen Adapter verwerfen.
Injektion:
Der Puls sollte vor und während der Injektion gemessen werden. Eine deutliche Erhöhung der Pulsfrequenz klingt normalerweise nach Verlangsamen oder Unterbrechen der Injektion schnell wieder ab.
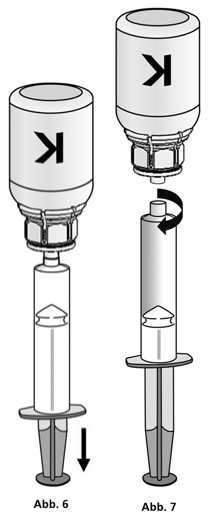
1. Die Spritze mit dem transparenten Adapter der Konzentratflasche verbinden. Die Flasche samt Einmalspritze umdrehen und das aufgelöste Präparat in die Spritze aufziehen (Abb. 6).
Die Injektionslösung in der Spritze sollte klar oder leicht schillernd sein. Nachdem das Produkt in die Spritze überführt wurde, den Spritzenzylinder fassen und die Spritze vom transparenten Adapter der Konzentratflasche entfernen (Abb. 7). Verwerfen Sie die Konzentratflasche mit dem Adapter.
c\j co
Vorgesehene Injektionsstelle mit einem Alkoholtupfer desinfizieren.
Die beigepackte Flügelkanüle auf die Spritze aufsetzen.
Stechen Sie die Flügelkanüle in die gewählte Vene. Wenn Sie die Vene vor der Punktion gestaut haben, damit Sie sie besser sehen können, müssen Sie die Stauung öffnen, bevor Sie mit der Injektion beginnen. Es darf kein Blut in die Spritze gelangen, da dies zur Bildung von Blutgerinnseln führen könnte.
5. Injizieren Sie die Lösung langsam in die Vene, wobei die Injektionsgeschwindigkeit höchstens 2 - 3 ml pro Minute betragen sollte.
Wenn Sie mehr als eine Flasche des Konzentrates benötigen, kann die Flügelkanüle in der Vene belassen werden. Zum Herstellen der gebrauchsfertigen Lösung immer ein neues Mix2Vial™-Set benutzen.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
7. INHABER DER ZULASSUNG
OCTAPHARMA GmbH Elisabeth-Selbert-Str. 11 40764 Langenfeld E-Mail: info@octapharma.de www.octapharma.de
8. ZULASSUNGSNUMMERN
Wilate 500: PEI.H.01918.03.1 Wilate 1000: PEI.H.01918.04.1
9. DATUM DER ZULASSUNG/VERLÄNGERUNG DER ZULASSUNG
Datum der Erteilung der Zulassung: 22. Dezember 2011 Datum der letzten Verlängerung der Zulassung: 29. Juli 2014
10. STAND DER INFORMATION
August 2014
11. VERSCHREIBUNGSSTATUS/APOTHEKENPFLICHT
Verschreibungspflichtig
12. SONSTIGE HINWEISE
Herkunftsland des Blutplasmas:
Deutschland, Estland, Finnland, Kroatien, Luxemburg, Norwegen, Österreich, Portugal, Schweden, Schweiz, Slowenien, Spanien, Tschechische Republik, Ungarn, USA.
Es wird auf die Dokumentationspflicht gemäß Transfusionsgesetz hingewiesen.
17/17
Bitte lesen Sie alle Anweisungen durch und befolgen Sie sie sorgfältig.
• Verwenden Sie Wilate nicht mehr nach dem Verfalldatum, das auf dem Etikett und dem Umkarton angegeben ist.
• Bitte achten Sie bei allen Arbeitsschritten strikt auf Hygiene.