Willfact 2000 I.e.
FACHINFORMATION
ZUSAMMENFASSUNG DER MERKMALE DES ARZNEIMITTELS
1. BEZEICHNUNG DES ARZNEIMITTELS
WILLFACT 2000 I.E.
Pulver und Lösungsmittel zur Herstellung einer Injektionslösung.
2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
WILLFACT liegt als Pulver und Lösungsmittel zur Herstellung einer Injektionslösung vor und enthält 2000 I.E. humanen von-Willebrand-Faktor je Durchstechflasche.
Nach Rekonstitution mit 20 ml Wasser für Injektionszwecke enthält WILLFACT 100 I.E. humanen von-Willebrand-Faktor je 1 ml rekonstituierter Lösung.
Bei Rekonstitution mit 20 ml Wasser für Injektionszwecke enthält das Arzneimittel etwa 100 I.E./ml humanen von-Willebrand-Faktor. Vor Hinzufügen von Albumin beträgt die spezifische Aktivität von WILLFACT 2000 I.E. mindestens 50 I.E. vWF:RCo/mg Protein.
Die von-Willebrand-Faktor-Aktivität (I.E.) wird im Vergleich zum internationalen Standard für von-Willebrand-Faktor-Konzentrat als Ristocetin-Cofaktor-Aktivität (vWF:RCo) gemessen.
Die Menge an humanem Faktor VIII in WILLFACT beträgt < 10 I.E./100 I.E. vWF:RCo. Zur Bestimmung der Stärke (I.E.) wird der chromogene Faktor-VIII-Gerinnungstest gemäß Europäischem Arzneibuch verwendet.
Die vollständige Auflistung der sonstigen Bestandteile siehe unter Abschnitt 6.1. Eine Durchstechflasche (2000 I.E.) WILLFACT enthält 0,6 mmol (13,8 mg) Natrium.
3. DARREICHUNGSFORM
Pulver und Lösungsmittel zur Herstellung einer Injektionslösung.
Das Pulver sollte weißes oder hellgelbes lyophilisiertes Pulver oder bröckelig, fest sein. Das Lösungsmittel sollte klar und farblos sein.
4. KLINISCHE ANGABEN
4.1. Anwendungsgebiete
WILLFACT ist indiziert zur Prävention und Therapie von Blutungen oder operationsbedingten Blutungen bei der von-Willebrand-Krankheit, wenn eine alleinige Therapie mit Desmopressin (DDAVP) unwirksam oder kontraindiziert ist.
WILLFACT darf nicht zur Therapie von Hämophilie A verwendet werden.
4.2. Dosierung, Art und Dauer der Anwendung
Die Therapie der von-Willebrand-Krankheit sollte von einem Arzt überwacht werden, der Erfahrung in der Behandlung von Gerinnungsstörungen hat.
Dosierung
Generell führt 1 I.E./kg von-Willebrand-Faktor zu einem Anstieg des zirkulierenden Spiegels von vWF:RCo um 0,02 I.E./ml (2 %).
Es sollten Spiegel von vWF:RCo von > 0,6 I.E./ml (60 %) und FVIIIC von > 0,4 I.E./ml (40 %) erreicht werden.
Eine Blutgerinnung kann nicht sichergestellt werden, bevor die blutgerinnungsfördernde Aktivität von FVIII (FVIII:C) 0,4 IE/ml (40 %) erreicht hat. Eine alleinige Injektion von von-Willebrand-Faktor führt erst nach mindestens 6-12 Stunden zu einem maximalen Anstieg von FVIII:C. Die alleinige Gabe von von-Willebrand-Faktor kann den FVIII:C-Spiegel nicht sofort korrigieren. Liegt der FVIII:C-Spiegel im Plasma des Patienten also unterhalb dieses kritischen Wertes, muss in allen Situationen, in denen eine rasche Korrektur der Blutgerinnung erforderlich ist, wie Therapie einer Blutung, schweres Trauma oder Notfalloperation, bei der ersten Injektion von von-Willebrand-Faktor gleichzeitig Faktor VIII injiziert werden, um einen blutungsstillend wirkenden Plasmaspiegel von FVIII:C zu erreichen.
Ist ein sofortiger Anstieg von FVIII:C jedoch nicht erforderlich, wie bei einer geplanten Operation, oder reicht der FVIII:C-Spiegel zu Beginn der Behandlung aus, um eine Blutstillung sicher zu stellen, kann der Arzt entscheiden, bei der ersten Injektion auf eine gleichzeitige Gabe von FVIII zu verzichten.
Therapiebeginn:
Zur Behandlung von Blutungen oder Verletzungen beträgt die erste WILLFACT-Dosis 40 bis 80 I.E./kg in Kombination mit der erforderlichen Menge des Faktor-VIII-Präparats und wird unmittelbar vor der Operation oder sobald wie möglich nach Einsetzen einer Blutung oder nach einem schweren Trauma verabreicht. Die erforderliche Menge des Faktor-VIII-Präparats wird anhand des Ausgangswerts des FVIII:C-Plasmaspiegels des Patienten so berechnet, dass ein entsprechender FVIII:C-Plasmaspiegel erreicht wird. Im Falle einer Operation sollte WILLFACT eine Stunde vor dem Eingriff verabreicht werden.
Eine anfängliche Dosis von 80 I.E./kg WILLFACT kann erforderlich sein, insbesondere bei Patienten mit der von-Willebrand-Krankheit Typ III, bei denen zum Erhalt ausreichender Spiegel höhere Dosen erforderlich sein können als bei den anderen Typen der von-Willebrand-Krankheit.
Bei geplanten Operationen sollte eine Therapie mit WILLFACT 12 bis 24 Stunden vor der Operation beginnen und 1 Stunde vor dem Eingriff wiederholt werden. Die gleichzeitige Anwendung eines Faktor-VIII-Präparats ist in diesem Fall nicht erforderlich, da das endogene FVIII:C normalerweise vor der Operation den kritischen Spiegel von 0,4 I.E./ml (40 %) erreicht hat. Dies sollte jedoch bei jedem Patienten individuell kontrolliert werden.
Falls erforderlich, sollte die Therapie mit einer geeigneten Dosis WILLFACT fortgesetzt werden, mit 40 bis 80 I.E./kg täglich in Form von einer oder zwei Injektionen über einen oder mehrere Tage. Die Dosis und Therapiedauer sind abhängig vom klinischen Zustand des Patienten, Art und Schweregrad der Blutung und dem vWF:RCo- sowie dem FVIII:C-Spiegel.
Langzeitprophylaxe
WILLFACT kann als langfristige Prophylaxe gegeben werden, in einer Dosis, die für jeden Patienten individuell festgelegt wird. WILLFACT-Dosen zwischen 40 und 60 I.E./kg, zwei- bis dreimal wöchentlich gegeben, reduzieren die Anzahl der Blutungsepisoden.
Zur Charakterisierung des Ansprechens von Kindern unter 6 Jahren auf die Behandlung mit WILLFACT liegen keine Daten aus klinischen Studien vor.
Die Anwendung von WILLFACT bei Kindern unter 12 Jahren ist nur in Einzelfallen dokumentiert; die Anwendung von WILLFACT bei Patienten, die noch nie mit von-Willebrand-Faktor behandelt wurden, ist in klinischen Studien nicht dokumentiert.
Art der Anwendung
Das Präparat wie unter 6.6 beschrieben auflösen. Das Arzneimittel wird intravenös mit einer maximalen Geschwindigkeit von 4 ml/Minute verabreicht.
4.3. Gegenanzeigen
Überempfindlichkeit gegen den Wirkstoff oder einen der sonstigen Bestandteile.
4.4. Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Bei aktiv blutenden Patienten wird empfohlen, das von-Willebrand-Faktor-Präparat mit einem niedrigen FVIII-Gehalt in Kombination mit einem FVIII-Präparat als Erstbehandlung zu verabreichen.
Wie bei jeder intravenösen Anwendung eines aus Plasma gewonnenen Proteins, sind
Überempfindlichkeitsreaktionen in Form einer Allergie möglich. Die Patienten müssen während der Injektion engmaschig überwacht und sorgfältig auf etwaige Symptome hin beobachtet werden. Die Patienten sollten über die Frühzeichen einer Überempfindlichkeit wie Quaddeln, generalisierte Urtikaria, Engegefühl in der Brust, Giemen, Hypotonie und Anaphylaxie informiert werden. Treten diese Symptome auf, muss die Anwendung sofort abgebrochen werden. Bei einem Schock müssen die geltenden Richtlinien zur Schocktherapie beachtet werden.
Standardmaßnahmen zur Verhinderung von Infektionen, die durch Anwendung von Arzneimitteln aus menschlichem Blut oder Plasma resultieren, sind unter anderem Auswahl der Spender, das Screening der Einzelspenden und der Plasmapools auf spezifische Infektionsmarker und die Aufnahme effektiver Herstellungsschritte zur Inaktivierung/Entfernung von Viren.
Dennoch lässt sich bei der Anwendung von Arzneimitteln, die aus menschlichem Blut oder Plasma gewonnen wurden, die Möglichkeit einer Übertragung infektiöser Agenzien nicht vollständig ausschließen. Das gilt auch für unbekannte oder neu auftretende Viren und andere Pathogene.
Die angewandten Maßnahmen werden für umhüllte Viren wie HIV, HBV und HCV als effektiv angesehen. Die angewandten Maßnahmen sind möglicherweise von nur begrenztem Wert gegen nicht umhüllte Viren wie HAV und Parvovirus B19. Eine Infektion durch den Parvovirus B19 kann für Schwangere (Infektion des Fötus) und Personen mit einer Immunschwäche oder erhöhter Erythropoese (z.B hämolytische Anämie) schwerwiegend sein.
Für Patienten, die regelmäßig Präparate aus menschlichem Blut oder Plasma erhalten, werden entsprechende Impfungen (Hepatitis A und Hepatitis B) empfohlen.
Es wird auf die Dokumentationspflicht hingewiesen, bei jeder Anwendung von WILLFACT Name und Chargennummer des Arzneimittels zu notieren, um einen Zusammenhang zwischen dem Patienten und der Chargennummer herstellen zu können.
Es besteht ein Risiko des Auftretens thrombotischer Ereignisse, insbesondere bei Patienten mit bekannten klinischen oder aus Untersuchungen der Laborwerte hervorgehenden Risikofaktoren. Risikopatienten müssen daher auf die Frühzeichen einer Thrombose hin überwacht werden. Eine Prophylaxe gegen venöse Thromboembolien muss nach den gegenwärtig geltenden Empfehlungen eingeleitet werden.
Bei der Anwendung eines FVIII-haltigen vWF-Präparats muss der behandelnde Arzt beachten, dass eine kontinuierliche Therapie zu einem exzessiven Anstieg von FVIII:C führen kann. Bei Patienten, die FVIII-haltige vWF-Präparate erhalten, müssen die Plasmaspiegel von FVIII:C überwacht werden, um anhaltend exzessive FVIII:C-Plasmaspiegel zu vermeiden, die das Risiko thrombotischer Ereignisse erhöhen können.
Patienten mit der von-Willebrand-Krankheit, insbesondere Patienten mit einer Erkrankung vom Typ III, können neutralisierende Antikörper (Inhibitoren) gegen vWF entwickeln. Wird der erwartete Plasmaspiegel der vWF:RCo-Aktivität nicht erreicht oder lässt sich die Blutung mit einer angemessenen Dosis nicht stoppen, sollte ein Assay durchgeführt werden, um zu bestimmen, ob ein vWF-Inhibitor vorliegt. Bei Patienten mit hohen Inhibitor-Spiegeln ist eine Therapie mit vWF möglicherweise nicht wirksam und es sollten andere Therapiemöglichkeiten erwogen werden.
Eine Durchstechflasche (2000 I.E.) WILLFACT enthält 0,6 mmol (13,8mg) Natrium. Wenn mehr als 3300 I.E. injiziert werden (mehr als 1 mmol Natrium), ist dies bei Patienten unter Natrium-kontrollierter Diät zu berücksichtigen.
4.5. Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Es sind keine Wechselwirkungen zwischen vWF-Präparaten und anderen Arzneimitteln bekannt.
4.6. Fertilität, Schwangerschaft und Stillzeit
Tierexperimentelle Studien sind unzureichend, um die Unbedenklichkeit bezüglich der Fertilität, Reproduktion, Schwangerschaft, embryonalen/fetalen Entwicklung oder peri- und postnatalen Entwicklung zu beurteilen.
Die Unbedenklichkeit von WILLFACT während der Schwangerschaft und Stillzeit wurde nicht in kontrollierten klinischen Studien untersucht.
Daher darf WILLFACT bei schwangeren und stillenden Frauen mit von-Willebrand-Faktor-Mangel nur bei strengster Indikationsstellung verwendet werden.
4.7. Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Es wurden keine Auswirkungen auf die Verkehrstüchtigkeit und das Bedienen von Maschinen beobachtet.
4.8. Nebenwirkungen
Obwohl die Sicherheit von WILLFACT als gut angesehen wird, könnten möglicherweise allergische oder anaphylaktische Reaktionen auftreten.
Bei der Bewertung nachfolgend angegebener Nebenwirkungen werden folgende Häufigkeitsangaben zugrunde gelegt:
Sehr häufig (> 1/10)
Häufig (> 1/100, < 1/10)
Gelegentlich (> 1/1.000, < 1/100)
Selten (> 1/10.000, < 1/1.000)
Sehr selten (< 1/10.000)
Nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar)
Erkrankungen des Immunsystems
Gelegentlich: Überempfindlichkeits- oder allergische Reaktionen. Diese können sich in einigen Fällen zu einer schweren Anaphylaxie weiterentwickeln (mit Schock).
Psychiatrische Erkrankungen Gelegentlich: Ruhelosigkeit
Erkrankungen des Nervensystems Gelegentlich: Kopfschmerzen, Kribbeln, Lethargie
Herzerkrankungen Gelegentlich: Tachykardie
Gefäßerkrankungen:
Gelegentlich: Hypotonie, Flush
Erkrankungen der Atemwege, des Brustraums und Mediastinums Gelegentlich: Giemen
Erkrankungen des Gastrointestinaltrakts Gelegentlich: Übelkeit, Erbrechen
Erkrankungen der Haut und des Unterhautzellgewebes Gelegentlich: Angioödem, generalisierte Urtikaria, Quaddeln
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Gelegentlich: Brennen und Stechen an der Applikationsstelle, Schüttelfrost, Engegefühl im Brustkorb Selten: Fieber
Untersuchungen
Sehr selten: Neutralisierende Antikörper (Inhibitoren) gegen vWF
Patienten mit der von-Willebrand-Krankheit, insbesondere Patienten mit einer Erkrankung vom Typ III, können sehr selten neutralisierende Antikörper (Inhibitoren) gegen vWF entwickeln. Mit vWF behandelte Patienten müssen sorgfältig mit Hilfe geeigneter klinischer Beobachtungen und Untersuchungen von Laborwerten auf das Auftreten von Inhibitoren hin überwacht werden. Treten derartige Inhibitoren auf, zeigt sich dies in Form eines unzureichenden klinischen Ansprechens. Derartige Antikörper präzipitieren und treten gemeinsam mit anaphylaktischen Reaktionen auf. Es wird empfohlen, in allen derartigen Fällen ein Hämophilie-Zentrum zu Rate zu ziehen. Daher sollten Patienten, die eine anaphylaktische Reaktion aufweisen, auf die Anwesenheit eines Inhibitors untersucht werden.
Nach Korrektur des von-Willebrand-Faktor-Mangels müssen aufgrund des Risikos einer Thrombose in gewissen Risikosituationen eine Überwachung auf Frühzeichen einer Thrombose oder disseminierten intravasalen Gerinnung und eine Prävention thromboembolischer Komplikationen entsprechend der gegenwärtigen Praxis vorgenommen werden.
Bei Patienten, die FVIII-haltige vWF-Präparate erhalten, kann durch anhaltend exzessive FVIII:C-Plasmaspiegel das Risiko thrombotischer Ereignisse erhöht sein.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung über: „Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel; Paul-Ehrlich-Institut, Paul-Ehrlich-Str. 5159, 63225 Langen; Tel: +49 6103 77 0; Fax: +49 6103 77 1234; Webseite: www.pei.de“ anzuzeigen.
4.9. Überdosierung
Es wurden keine Fälle einer Überdosierung von WILLFACT berichtet.
Bei einer erheblichen Überdosierung kann es zu thromboembolischen Ereignissen kommen.
5. PHARMAKOLOGISCHE EIGENSCHAFTEN
5.1. Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Antihämorrhagika: von-Willebrand-Faktor ATC-Code: B02BD10
WILLFACT verhält sich wie körpereigener von-Willebrand-Faktor.
Die Gabe des von-Willebrand-Faktors ermöglicht die Korrektur von Blutgerinnungsstörungen bei Patienten mit Mangel an diesem Faktor. Das Präparat hat zwei Wirkungsweisen:
• vWF stellt die Thrombozytenadhäsion am Subendothel des Gefäßes am Ort der Gefäßschädigung wieder her (da er sich sowohl an das Subendothel des Gefäßes als auch an die Thrombozytenmembran bindet) und führt so zu einer primären Blutgerinnung. Das zeigt sich in einer Verkürzung der Blutungszeit. Diese Wirkung hängt bekanntermaßen erheblich vom Grad der Multimerisation des aktiven Bestandteils ab.
• Von-Willebrand-Faktor bewirkt eine verzögerte Korrektur des ebenfalls vorliegenden Faktor-VIII-Mangels. Bei intravenöser Verabreichung bindet sich von-Willebrand-Faktor an endogenen Faktor VIII (der normalerweise vom Patienten selbst hergestellt wird). Durch Stabilisierung dieses Faktors verhindert er dessen raschen Abbau. Aufgrund dessen führt die Anwendung von reinem vWF (vWF-Präparat mit niedrigem FVIII-Spiegel) zu einem Anstieg des FVIII:C-Spiegels auf Normalwerte als sekundäre Wirkung nach der ersten Infusion. Bei Anwendung eines FVIII:C-haltigen vWF-Präparats kommt es unmittelbar nach der ersten Infusion zu einem Anstieg des FVIII:C-Spiegels auf Normalwerte.
5.2. Pharmakokinetische Eigenschaften
Eine pharmakokinetische Studie mit WILLFACT wurde bei 8 Patienten mit der von-Willebrand-Krankheit Typ III durchgeführt. Es zeigte sich, dass für vWF:RCo:
• die mittlere AUC0.^ nach Einmalgabe von 100 I.E./kg WILLFACT 3444 I.E. Xh/dl beträgt,
• der maximale Plasmaspiegel zwischen 30 Minuten und 1 Stunde nach der Injektion erreicht wird,
• die durchschnittliche Recovery 2,1 [I.E./dl]/[I.E./kg] des injizierten Präparats beträgt,
• die Halbwertzeit zwischen 8 und 14 Stunden liegt, mit einem Durchschnittswert von 12 Stunden,
• die mittlere Clearance 3,0 ml/h/kg beträgt.
Der Anstieg des FVIII-Spiegels auf Normalwerte verläuft progredient und variabel und erfolgt in der Regel nach 6 bis 12 Stunden. Diese Wirkung hält 2 bis 3 Tage an.
Der Anstieg des FVIII-Spiegels verläuft progredient, der Spiegel erreicht nach 6 bis 12 Stunden wieder Normalwerte. Der FVIII-Spiegel erhöht sich um durchschnittlich 6 % (I.E./dl) pro Stunde. Somit steigt selbst bei Patienten mit einem FVIII:C-Spiegel, der zu Beginn unter 5 % (I.E./dl) beträgt, der FVIII:C-Spiegel 6 Stunden nach der Injektion auf etwa 40 % (I.E./dl) an, dieser Spiegel bleibt über 24 Stunden erhalten.
5.3. Präklinische Daten zur Sicherheit
Basierend auf den Daten von verschiedenen präklinischen Studien am Tiermodell gibt es keine Hinweise auf andere toxische Effekte von WILLFACT außer auf die immunogene Aktivität der menschlichen Proteine in Labortieren. Tierversuche mit wiederholten Gaben sind aufgrund der sich bildenden Antikörper auf Fremdproteine nicht sinnvoll durchführbar.
Die präklinischen Daten zur Sicherheit weisen nicht darauf hin, dass WILLFACT mutagenes Potenzial besitzt.
6. PHARMAZEUTISCHE ANGABEN 6.1. Liste der sonstigen Bestandteile
Pulver: Human-Albumin, Argininhydrochlorid, Glycin, Natriumcitrat und Calciumchlorid-Dihydrat. Lösungsmittel: Wasser für Injektionszwecke.
6.2. Inkompatibilitäten
WILLFACT darf nicht mit anderen Arzneimitteln gemischt werden, außer mit aus Plasma gewonnenem Gerinnungsfaktor VIII, der von LFB-BIOMEDICAMENTS hergestellt wird und mit welchem eine Kompatibilitätsstudie durchgeführt wurde. Dieser Gerinnungsfaktor VIII ist allerdings nicht in allen europäischen Ländern in Verkehr.
Es dürfen nur zugelassene Injektionssets aus Polypropylen verwendet werden, da es infolge einer Adsorption von humanem von-Willebrand-Faktor an der inneren Oberfläche einiger Injektionsmaterialien zu einem Therapieversagen kommen kann.
6.3. Dauer der Haltbarkeit
3 Jahre.
Die chemische und physikalische Stabilität der gebrauchsfertigen Lösung wurde für 24 Stunden bei 25°C nachgewiesen.
Aus mikrobiologischer Sicht sollte das Arzneimittel sofort verwendet werden. Falls die Lösung nicht sofort verabreicht wird, fallen die Lagerungszeiten der gebrauchsfertigen Lösung und die Lagerungsbedingungen vor der Anwendung in den Verantwortungsbereich des Anwenders.
6.4. Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Nicht über 25°C lagern. In der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen. Nicht einfrieren.
6.5. Art und Inhalt des Behältnisses
1 Packung enthält: Pulver in einer Durchstechflasche (Typ-I-Glas) mit einem Brombutylstopfen + 20 ml Lösungsmittel in einer Durchstechflasche (Typ-I-Glas) mit einem Chlorbutylstopfen und mit Transfersystem.
6.6. Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung
Rekonstitution:
Die gegenwärtig geltenden Richtlinien für aseptische Verfahren sind einzuhalten. Das Transfersystem wird, wie unten beschrieben, nur zur Rekonstitution des Arzneimittels verwendet. Es ist nicht für die Verabreichung des Arzneimittels an den Patienten gedacht.


• Erwärmen Sie die beiden Durchstechflaschen (Pulver und Lösungsmittel) auf eine Temperatur von maximal 25 °C.
• Entfernen Sie die Schutzkappen von der Durchstechflasche mit Lösungsmittel (Wasser für Injektionszwecke) und der Durchstechflasche mit Pulver.
• Desinfizieren Sie die Oberfläche beider Stopfens.
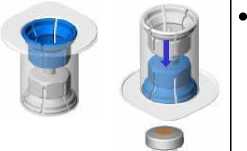
Entfernen Sie die Kappe vom Mix2Vial-Set. Befestigen Sie das Mix2Vial-Set mit dem blauen Adapter am Stopfen der Durchstechflasche mit Lösungsmittel ohne das Set aus seiner Verpackung zu nehmen,.


• Entfernen Sie die Verpackung und werfen Sie sie weg. Achten Sie darauf, den nun frei gewordenen Teil des Sets nicht zu berühren.
• Drehen Sie die Lösungsmittelflasche mit dem aufgesetzten Set um und befestigen Sie dieses mit dem transparenten Adapter an der Durchstechflasche mit Pulver. Das Lösungsmittel fließt automatisch in die Durchstechflasche mit Pulver. Halten Sie die beiden Flaschen und schwenken Sie sie vorsichtig, bis das Produkt vollständig gelöst ist.

Das Pulver löst sich im Allgemeinen schnell und sollte sich in weniger als 10 Minuten vollständig gelöst haben.
Die gebrauchsfertige Lösung sollte klar oder leicht opaleszierend, farblos oder leicht gelblich sein.
Anwendung:

• Halten Sie die Durchstechflasche mit dem rekonstituierten Produkt senkrecht, während Sie eine sterile Spritze mit dem Mix2Vial-Adapter verbinden. Danach ziehen Sie das Präparat langsam in die Spritze auf.
• Wenn das Präparat in die Spritze aufgezogen ist, halten Sie die Spritze (mit dem Kolben nach unten gerichtet) gut fest, schrauben den Mix2Vial-Adapter ab und ersetzen ihn durch eine intravenöse Nadel oder Flügelkanüle.
• Drücken Sie die Luft aus der Spritze und stechen Sie die Nadel nach Desinfektion der Punktionsstelle in die gewählte Vene.
• Injizieren Sie die Lösung unmittelbar nach der Rekonstitution langsam als Einzeldosis intravenös mit einer Höchstgeschwindigkeit von 4 ml/Minute.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
7. INHABER DER ZULASSUNG
LFB-BIOMEDICAMENTS 3, avenue des Tropiques,
BP 305 - LES ULIS,
91958 Courtabreuf Cedex FRANKREICH
8. ZULASSUNGSNUMMER
PEI.H.03530.03.1
9. DATUM DER ERTEILUNG DER ZULASSUNG/VERLÄNGERUNG DER ZULASSUNG
Datum der Erteilung der Zulassung 15 Mai 2009
Datum der letzten Verlängerung der Zulassung {TT. Monat JJJJ}
10. STAND DER INFORMATION
Februar 2015
11. VERSCHREIBUNGSSTATUS/APOTHEKENPFLICHT
Verschreibungspflichtig
12. HERKUNFTSLÄNDER DES VERWENDETEN BLUTPLASMAS
Deutschland, Österreich und USA