Zoledronsäure Al 4 Mg/100 Ml Infusionslösung
Fachinformation (Zusammenfassung der Merkmale des Arzneimittels/SPC)
1. Bezeichnung des Arzneimittels
Zoledronsäure AL 4 mg/100 ml Infusionslösung
2. Qualitative und quantitative Zusammensetzung
1 Durchstechflasche mit 100 ml Infusionslösung enthält 4 mg Zoledronsäure, entsprechend 4,26 mg Zoledronsäure 1 H2O.
Sonstiger Bestandteil mit bekannter Wirkung: 1 Durchstechflasche enthält Natriumcitrat, aber weniger als 1 mmol (23 mg) Natrium pro Durchstechflasche, d.h., es ist nahezu „natriumfrei“.
Vollständige Auflistung der sonstigen Bestandteile siehe Abschnitt 6.1.
3. Darreichungsform
Infusionslösung
Klare, farblose Lösung, frei von sichtbaren Partikeln.
pH-Wert: 5,5-7,0 Osmolarität: 0,27-0,33 Osmol/kg
4. Klinische Angaben
4.1 Anwendungsgebiete
• Prävention skelettbezogener Komplikationen (pathologische Frakturen, Wirbelkompressionen, Bestrahlung oder Operation am Knochen oder tumorinduzierte Hyperkalzämie) bei erwachsenen Patienten mit fortgeschrittenen, auf das Skelett ausgedehnten Tumorerkrankungen.
• Behandlung erwachsener Patienten mit tumorinduzierter Hyperkalzämie (TIH).
4.2 Dosierung und Art der Anwendung
Zoledronsäure AL darf Patienten nur von Ärzten mit Erfahrung bei der
Anwendung von intravenösen Bisphosphonaten verschrieben und verabreicht
werden. An Patienten, die mit Zoledronsäure AL behandelt werden, sollen die
Packungsbeilage und die Erinnerungskarte für Patienten ausgehändigt werden.
Dosierung
Prävention skelettbezogener Komplikationen bei Patienten mit fortgeschrittenen, auf das Skelett ausgedehnten Tumorerkrankungen Erwachsene und ältere Patienten
Die empfohlene Dosis zur Prävention skelettbezogener Ereignisse bei Patienten mit fortgeschrittenen, auf das Skelett ausgedehnten Tumorerkrankungen beträgt 4 mg Zoledronsäure in Abständen von 3-4 Wochen.
Diese Patienten sollten zusätzlich 500 mg Calcium und 400 I.E. Vitamin D pro Tag oral erhalten.
Bei der Entscheidung, Patienten mit Knochenmetastasen zur Prävention skelettbezogener Komplikationen zu behandeln, sollte berücksichtigt werden, dass die Wirkung nach 2-3 Monaten eintritt.
Behandlung der TIH Erwachsene und ältere Patienten
Die empfohlene Dosierung bei Hyperkalzämie (Albumin-korrigierter SerumCalcium-Spiegel >12,0 mg/dl oder 3,0 mmol/l) beträgt eine Einzeldosis 4 mg Zoledronsäure.
Nierenfunktionsstörungen
TIH
Die Behandlung mit Zoledronsäure AL bei Patienten mit TIH und einer schweren Nierenfunktionsstörung sollte nur nach vorheriger Nutzen-RisikoBeurteilung der Behandlung erwogen werden. In den klinischen Studien waren Patienten mit einem Serum-Kreatinin >400 gmol/l oder >4,5 mg/dl ausgeschlossen. Bei Patienten mit TIH und einem Serum-Kreatinin <400 gmol/l oder <4,5 mg/dl sind keine Dosisanpassungen erforderlich (siehe Abschnitt 4.4).
Prävention skelettbezogener Komplikationen bei Patienten mit fortgeschrittenen, auf das Skelett ausgedehnten Tumorerkrankungen Zu Beginn der Behandlung mit Zoledronsäure AL sollte bei Patienten mit multiplem Myelom oder metastatischen Knochenläsionen aufgrund solider Tumoren das Serum-Kreatinin und die Kreatinin-Clearance (CrCl) bestimmt werden. Die CrCl wird aus dem Serum-Kreatinin unter Verwendung der Cockcroft-Gault-Formel berechnet. Bei Patienten, die bereits vor Beginn der Behandlung eine schwere Nierenfunktionsstörung aufweisen, die für diese Patientenpopulation als CrCl <30 ml/min definiert ist, wird Zoledronsäure AL nicht empfohlen. In den klinischen Studien mit Zoledronsäure waren Patienten mit einem Serum-Kreatinin >265 gmol/l oder >3,0 mg/dl ausgeschlossen.
Bei Patienten mit normaler Nierenfunktion (definiert als CrCl >60 ml/min) kann Zoledronsäure 4 mg/100 ml Infusionslösung direkt und ohne weitere Vorbereitung gegeben werden. Bei Patienten mit Knochenmetastasen, die vor Beginn der Therapie eine leichte bis mittelschwere Nierenfunktionsstörung aufweisen, die bei dieser Patientenpopulation als CrCl 30-60 ml/min definiert ist, werden verringerte Dosierungen von Zoledronsäure AL empfohlen (siehe auch Abschnitt 4.4):
|
Kreatinin-Clearance zu Beginn der Behandlung (ml/min) |
Empfohlene Zoledronsäure-Dosierung* |
|
>60 |
4,0 mg Zoledronsäure |
|
50-60 |
3,5 mg* Zoledronsäure |
|
40-49 |
3,3 mg* Zoledronsäure |
|
30-39 |
3,0 mg* Zoledronsäure |
• Die Dosierungen wurden berechnet unter Annahme einer Ziel-AUC von 0,66 (mgWl) (CrCl = 75 ml/min). Die verminderten Dosen für Patienten mit Nierenfunktionsstörungen lassen erwarten, dass die gleiche AUC erreicht wird, wie sie bei Patienten mit einer Kreatinin-Clearance von 75 ml/min beobachtet wurde.
Nach Beginn der Behandlung sollte vor jeder Gabe von Zoledronsäure AL das Serum-Kreatinin gemessen und auf die weitere Behandlung verzichtet werden, wenn sich die Nierenfunktion verschlechtert hat. In den klinischen Studien wurde eine Verschlechterung der Nierenfunktion wie folgt definiert:
• Bei Patienten mit normalem Serum-Kreatinin zu Beginn der Behandlung (<1,4 mg/dl oder <124 pmol/l) ein Anstieg um 0,5 mg/dl oder 44 pmol/l.
• Bei Patienten mit erhöhtem Serum-Kreatinin zu Beginn der Behandlung (>1,4 mg/dl oder >124 pmol/l) ein Anstieg um 1,0 mg/dl oder 88 pmol/l.
In klinischen Studien wurde die Behandlung mit Zoledronsäure erst dann erneut aufgenommen, wenn die Kreatinin-Werte nur noch maximal 10% über dem Ausgangswert lagen (siehe Abschnitt 4.4). Die Therapie mit Zoledronsäure AL sollte mit der gleichen Dosis wie vor der Unterbrechung der Behandlung wieder aufgenommen werden.
Kinder und Jugendliche
Die Unbedenklichkeit und Wirksamkeit von Zoledronsäure bei Kindern im Alter von 1 bis 17 Jahren ist nicht nachgewiesen. Zurzeit vorliegende Daten sind in Abschnitt 5.1 beschrieben; eine Dosierungsempfehlung kann jedoch nicht gegeben werden.
Art der Anwendung
Intravenöse Anwendung.
Zoledronsäure AL 4 mg/100 ml Infusionslösung sollte auf einmal als intravenöse Infusion über mindestens 15 Minuten gegeben werden.
Bei Patienten mit normaler Nierenfunktion, definiert als CrCl >60ml/min, darf die Zoledronsäure 4 mg/100 ml Infusionslösung nicht weiter verdünnt werden.
Bei Patienten mit leichter bis mittelschwerer Nierenfunktionsstörung werden verringerte Dosen von Zoledronsäure empfohlen (siehe oben Abschnitt „Dosierung" und Abschnitt 4.4).
Zur Herstellung einer verringerten Dosis für Patienten mit einer CrCl <60 ml/min wird auf nachfolgende Tabelle 1 verwiesen. Entnehmen Sie das angegebene Volumen Zoledronsäure-Infusionslösung aus der Flasche und ersetzen Sie es durch das gleiche Volumen einer sterilen 0,9%igen Natriumchloridlösung oder einer 5%igen Glucoselösung.
Tabelle 1: Herstellung verringerter Dosierungen von Zoledronsäure 4 mg/100 ml Infusionslösung
|
Kreatinin-Clearance (ml/min) zu Beginn der Behandlung |
Entnehmen Sie die folgende Menge von Zoledronsäure-Infusionslösung (ml) |
Ersetzen Sie diese mit dem folgenden Volumen einer sterilen 0,9%igen Natriumchloridlösun g oder 5%igen Glucoselösung (ml) |
Geänderte Dosierung (mg Zoledronsäure in 100 ml) |
|
50-60 |
12,0 |
12,0 |
3,5 |
|
40-49 |
18,0 |
18,0 |
3,3 |
|
30-39 |
25,0 |
25,0 |
3,0 |
Die Zoledronsäure AL 4 mg/100 ml Infusionslösung darf nicht mit anderen Infusionslösungen gemischt werden und sollte als gesonderte intravenöse Lösung über eine eigene Infusionslinie gegeben werden.
Vor und nach der Gabe von Zoledronsäure AL müssen die Patienten ausreichend hydratisiert sein.
4.3 Gegenanzeigen
• Überempfindlichkeit gegen den Wirkstoff, andere Bisphosphonate oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile
• Stillen (siehe Abschnitt 4.6).
4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Allgemein
Vor der Applikation von Zoledronsäure AL muss eingeschätzt werden, ob die Patienten in einem adäquaten Hydratationszustand sind.
Eine Hyperhydratation ist bei Patienten mit einem Risiko für eine Herzinsuffizienz zu vermeiden.
Die üblicherweise mit einer Hyperkalzämie in Zusammenhang stehenden metabolischen Parameter wie z.B. die Serumspiegel von Calcium, Phosphat und Magnesium sollten nach Einleitung der Therapie mit Zoledronsäure sorgfältig überwacht werden. Bei Auftreten von Hypokalzämie, Hypophosphatämie oder Hypomagnesiämie kann eine kurzzeitige Substitution notwendig werden. Unbehandelte Patienten mit Hyperkalzämie weisen im
Allgemeinen eine Nierenfunktionsstörung auf. Deshalb sollte für eine sorgfältige Überwachung der Nierenfunktion gesorgt werden.
Zoledronsäure AL 4 mg/100 ml Infusionslösung enthält denselben Wirkstoff wie Zoledronsäure 5 mg/100 ml. Patienten, die mit Zoledronsäure AL behandelt werden, sollten nicht gleichzeitig mit Zoledronsäure 5 mg/100 ml oder anderen Bisphosphonaten behandelt werden, weil die kombinierte Wirkung dieser Stoffe nicht bekannt ist.
Niereninsuffizienz
Bei Patienten mit TIH und Hinweisen auf eine Verschlechterung der Nierenfunktion ist darauf zu achten, dass der potenzielle Nutzen einer Behandlung mit Zoledronsäure AL gegenüber möglichen Risiken überwiegt.
Bei der Entscheidung zur Behandlung von Patienten mit Knochenmetastasen zur Prävention skelettbezogener Ereignisse sollte berücksichtigt werden, dass der Behandlungseffekt nach 2-3 Monaten einsetzt.
Zoledronsäure wurde mit Berichten von Nierenfunktionsstörungen in Zusammenhang gebracht. Faktoren, die die Wahrscheinlichkeit einer Verschlechterung der Nierenfunktion erhöhen können, sind unter anderem Dehydratation, vorbestehende Nierenfunktionsstörungen, mehrere Behandlungszyklen mit Zoledronsäure und anderen Bisphosphonaten sowie die Anwendung anderer nephrotoxischer Arzneimittel. Auch wenn das Risiko bei einer Dosierung von 4 mg Zoledronsäure, gegeben über 15 Minuten, verringert ist, kann dennoch eine Verschlechterung der Nierenfunktion auftreten. Über eine Verschlechterung der Nierenfunktion, ein Fortschreiten bis hin zum Nierenversagen und zur Dialyse wurde bei Patienten nach der Initialdosis oder nach einmaliger Dosis von 4 mg Zoledronsäure berichtet. Ein Anstieg des Serum-Kreatinins tritt bei einigen Patienten auch unter chronischer Anwendung von Zoledronsäure in der empfohlenen Dosis zur Prävention skelettbezogener Ereignisse auf, wenngleich weniger häufig.
Vor jeder Gabe von Zoledronsäure AL sollten die Serum-Kreatinin-Werte der Patienten bestimmt werden. Zu Beginn der Behandlung von Patienten mit Knochenmetastasen mit leichten und mittelschweren Nierenfunktionsstörungen werden niedrigere Dosen von Zoledronsäure empfohlen. Bei Hinweis auf eine Verschlechterung der Nierenfunktion während der Behandlung sollte Zoledronsäure AL abgesetzt werden. Zoledronsäure AL sollte erst dann erneut gegeben werden, wenn die Serum-Kreatinin-Werte nur noch maximal 10% über dem Ausgangswert liegen (siehe Abschnitt 4.2). Die Behandlung mit Zoledronsäure AL sollte mit der gleichen Dosierung wie vor der Behandlungsunterbrechung wieder aufgenommen werden.
Angesichts eines möglichen Einflusses von Zoledronsäure auf die Nierenfunktion, kann wegen des Fehlens von Daten zur klinischen Verträglichkeit bei Patienten mit schweren Nierenfunktionsstörungen zu Beginn
der Behandlung (in klinischen Studien definiert als Serum-Kreatinin >400 gmol/l oder >4,5 mg/dl bei Patienten mit TIH bzw. >265 gmol/l oder >3,0 mg/dl bei Patienten mit Tumoren und Knochenmetastasen) sowie nur begrenzter pharmakokinetischer Daten bei Patienten mit schweren Nierenfunktionsstörungen zu Beginn der Behandlung (Kreatinin-Clearance <30 ml/min) die Anwendung von Zoledronsäure bei Patienten mit schweren Nierenfunktionsstörungen nicht empfohlen werden.
Leberinsuffizienz
Da für die Behandlung von Patienten mit schweren Leberfunktionsstörungen nur wenige klinische Daten verfügbar sind, können für diese Patienten keine speziellen Empfehlungen gegeben werden.
Osteonekrosen im Kieferbereich
Über Osteonekrosen im Kieferbereich wurde bei Patienten berichtet. Dies betraf in erster Linie Tumorpatienten, die mit Arzneimitteln, die die Knochenresorption hemmen, wie Zoledronsäure, behandelt wurden. Viele dieser Patienten erhielten zusätzlich eine Chemotherapie und Kortikosteroide. Die Mehrzahl der berichteten Fälle trat bei gleichzeitiger dentaler Behandlung wie z.B. Zahnextraktion auf. Viele Patienten hatten Anzeichen einer lokalen Infektion einschließlich Osteomyelitis.
Vor der Behandlung mit Bisphosphonaten sollte bei Patienten mit gleichzeitig vorhandenen Risikofaktoren (z.B. Tumoren, Chemotherapie, Kortikosteroide, mangelhafte Mundhygiene) eine zahnärztliche Untersuchung mit angemessenen prophylaktischen zahnmedizinischen Maßnahmen erwogen werden.
Der Beginn der Behandlung oder eines neuen Behandlungszyklus sollte bei Patienten mit nicht- verheilten, offenen Weichteilläsionen im Mund, außer in medizinischen Notfallsituationen, verschoben werden. Eine zahnärztliche Untersuchung mit angemessener präventiver Zahnbehandlung und eine individuelle Nutzen-Risiko-Bewertung werden vor der Behandlung mit Bisphosphonaten bei Patienten mit begleitenden Risikofaktoren empfohlen.
Die folgenden Risikofaktoren sollten in Betracht gezogen werden, wenn das individuelle Risiko für das Auftreten einer ONJ bestimmt wird:
• Potenz des Bisphosphonats (höheres Risiko für hoch potente Substanzen), Art der Anwendung (höheres Risiko bei parenteraler Anwendung) und kumulative Bisphosphonat-Dosis,
• Krebs, Begleiterkrankungen (z.B. Anämie, Koagulopathien, Infektion, Rauchen,
• begleitende Therapien: Chemotherapie, Angiogenese-Inhibitoren (siehe Abschnitt 4.5), Strahlentherapie an Kopf und Hals, Kortikosteroide,
• Zahnerkrankungen in der Vorgeschichte, mangelhafte Mundhygiene, periodontale Erkrankung, invasive Zahnbehandlungen (z.B. Zahnextraktionen) und schlecht sitzende Zahnprothese.
Während der Behandlung sollten bei diesen Patienten invasive dentale Eingriffe möglichst vermieden werden. Bei Patienten, bei denen während der Behandlung mit Bisphosphonaten eine Osteonekrose im Kieferbereich auftritt, kann ein dentaler Eingriff zur Verschlechterung des Zustandes führen. Für Patienten, bei denen invasive dentale Eingriffe erforderlich sind, gibt es keine Daten, die darauf hinweisen, ob eine Unterbrechung der BisphosphonatBehandlung das Risiko einer Osteonekrose im Kieferbereich vermindert. Für den Behandlungsplan eines jeden Patienten sollte die klinische Beurteilung des behandelnden Arztes, basierend auf der individuellen Nutzen-Risiko-Abwägung, ausschlaggebend sein.
Der Behandlungsplan für Patienten, die eine Osteonekrose im Kieferbereich entwickeln, sollte in enger Zusammenarbeit zwischen dem behandelnden Arzt und einem Zahnarzt oder Kieferchirurgen mit Expertise bei der Behandlung von Kieferosteonekrosen erstellt werden. Eine vorübergehende Unterbrechung der Zoledronsäure-Behandlung sollte in Erwägung gezogen werden, bis der Zustand behoben ist und die dazu beitragenden Risikofaktoren soweit möglich begrenzt werden können.
Knochennekrose des äußeren Gehörgangs
Bei der Anwendung von Bisphosphonaten wurde über Knochennekrosen des äußeren Gehörgangs berichtet, und zwar hauptsächlich im Zusammenhang mit Langzeitbehandlungen. Zu den möglichen Risikofaktoren für eine Knochennekrose des äußeren Gehörgangs zählen die Anwendung von Steroiden und chemotherapeutischen Behandlungen und/oder lokale Risikofaktoren wie z.B. Infektionen oder Traumata. Die Möglichkeit einer Knochennekrose des äußeren Gehörgangs sollte bei Patienten, die Bisphosphonate erhalten und mit Ohrsymptomen, einschließlich chronischer Ohreninfektionen, vorstellig werden, in Betracht gezogen werden.
Muskel- und Skelettschmerzen
Nach der Markteinführung wurde über starke und gelegentlich zur Einschränkung der Beweglichkeit führende Knochen-, Gelenk- und/oder Muskelschmerzen berichtet bei Patienten, die Zoledronsäure angewendet haben. Diese Berichte waren jedoch selten. Der Zeitpunkt des Auftretens der Symptome variierte vom ersten Tag nach Beginn der Behandlung bis zu mehreren Monaten später. Bei den meisten Patienten besserten sich die Symptome nach Beendigung der Behandlung. Bei einem Teil der Patienten traten die Symptome nach Reexposition mit Zoledronsäure oder einem anderen Bisphosphonat wieder auf.
Atypische Femurfrakturen
Atypische subtrochantäre und diaphysäre Femurfrakturen wurden unter Bisphosphonat-Therapie berichtet, vor allem bei Patienten unter Langzeitbehandlung gegen Osteoporose. Diese transversalen oder kurzen Schrägfrakturen können überall entlang des Oberschenkelknochens auftreten, direkt unterhalb des Trochanter minor bis direkt oberhalb der Femurkondylen. Diese Frakturen entstehen nach einem minimalen Trauma oder ohne Trauma und manche Patienten verspüren Oberschenkel- oder Leistenschmerzen, oft im Zusammenhang mit Anzeichen einer Niedrig-Energie-Fraktur in bildgebenden Verfahren Wochen bis Monate vor dem Auftreten einer manifesten Femurfraktur. Frakturen treten häufig bilateral auf. Aus diesem Grund sollte bei Patienten, die mit Bisphosphonaten behandelt werden und eine Femurschaftfraktur hatten, der kontralaterale Femur ebenfalls untersucht werden. Über eine schlechte Heilung dieser Frakturen ist ebenfalls berichtet worden. Bei Patienten mit Verdacht auf eine atypische Femurfraktur sollte ein Absetzen der Bisphosphonat-Therapie, vorbehaltlich einer Beurteilung des Patienten, auf Grundlage einer individuellen Nutzen-Risiko-Bewertung in Betracht gezogen werden.
Während einer Behandlung mit Bisphosphonaten sollte den Patienten geraten werden, über jegliche Oberschenkel-, Hüft- oder Leistenschmerzen zu berichten, und jeder Patient mit diesen Symptomen sollte auf eine unvollständige Femurfraktur hin untersucht werden.
Hypokalzämie
Hypokalzämie wurde bei mit Zoledronsäure behandelten Patienten berichtet. Herzrhythmusstörungen und neurologische Nebenwirkungen (einschließlich Konvulsionen, Hypästhesie und Tetanie) wurden als Folge von Fällen einer schweren Hypokalzämie berichtet. Fälle von schwerer Hypokalzämie, die eine Hospitalisierung erforderten, wurden berichtet. In einigen Fällen kann eine Hypokalzämie lebensbedrohlich sein (siehe Abschnitt 4.8). Vorsicht ist geboten, wenn Zoledronsäure zusammen mit anderen Arzneimitteln angewendet wird, von denen bekannt ist, dass sie eine Hypokalzämie verursachen. Diese könnten einen synergistischen Effekt haben, der zu einer schweren Hypokalzämie führt (siehe Abschnitt 4.5). Das Serumcalcium muss bestimmt werden und eine Hypokalzämie muss vor Beginn der Therapie mit Zoledronsäure korrigiert werden. Die Patienten müssen angemessen mit Calcium und Vitamin D versorgt werden.
Natrium
Dieses Arzneimittel enthält Natrium, aber weniger als 1 mmol (23 mg) Natrium pro Infusionsflasche, d.h., es ist nahezu „natriumfrei“.
4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
In klinischen Studien wurde Zoledronsäure gemeinsam mit häufig verwendeten antitumorösen Arzneimitteln sowie mit Diuretika, Antibiotika und Analgetika angewandt, ohne dass klinisch erkennbare Wechselwirkungen aufgetreten wären. Zoledronsäure wird nur unwesentlich an Plasmaproteine gebunden und hemmt in vitro keine humanen P450-Enzyme (siehe Abschnitt 5.2). Spezielle klinische Studien zu Wechselwirkungen wurden jedoch nicht durchgeführt.
Vorsicht ist geboten, wenn Bisphosphonate gleichzeitig mit Aminoglykosiden, Calcitonin oder Schleifendiuretika angewendet werden, weil diese Substanzklassen einen additiven Effekt zeigen können, der zu einem niedrigeren Serum-Calcium-Spiegel über einen längeren Zeitraum als erforderlich führen kann (siehe Abschnitt 4.4).
Vorsicht ist geboten, wenn Zoledronsäure zusammen mit anderen Arzneimitteln gegeben wird, die möglicherweise ebenfalls die Nierenfunktion beeinträchtigen könnten. Es ist auch auf eine möglicherweise während der Behandlung auftretende Hypomagnesiämie zu achten.
Bei Patienten mit Multiplem Myelom kann das Risiko für eine Verschlechterung der Nierenfunktion erhöht sein, wenn Zoledronsäure zusammen mit Thalidomid angewendet wird.
Vorsicht ist geboten, wenn Zoledronsäure zusammen mit anti-angiogenetischen Arzneimitteln angewendet wird, da eine erhöhte Inzidenz von Osteonekrosen im Kieferbereich (ONJ) bei Patienten beobachtet wurde, die gleichzeitig mit diesen Arzneimitteln behandelt wurden.
4.6 Fertilität, Schwangerschaft und Stillzeit
Schwangerschaft
Es liegen keine hinreichenden Daten über die Verwendung von Zoledronsäure bei Schwangeren vor. Tierexperimentelle Studien mit Zoledronsäure haben eine Reproduktionstoxizität gezeigt (siehe Abschnitt 5.3). Das potenzielle Risiko für den Menschen ist nicht bekannt. Zoledronsäure sollte nicht während der Schwangerschaft angewendet werden. Frauen im gebärfähigen Alter sollte empfohlen werden, eine Schwangerschaft zu vermeiden.
Stillzeit
Es ist nicht bekannt, ob Zoledronsäure in die Muttermilch übergeht. Zoledronsäure ist in der Stillzeit kontraindiziert (siehe Abschnitt 4.3).
Fertilität
Zoledronsäure wurde bei Ratten hinsichtlich möglicher unerwünschter Wirkungen auf die Fertilität der Eltern- und der F1-Generation untersucht. Dabei kam es zu einem übersteigerten pharmakologischen Effekt, der auf die hemmende Wirkung der Substanz auf den Calciummetabolismus im Knochen zurückgeführt wurde. Dies führte zu peripartaler Hypokalzämie, einem Klasseneffekt von Bisphosphonaten, Dystokie und einer frühzeitigen Beendigung der Studie. Die Ergebnisse lassen daher keinen definitiven Schluss auf die Wirkung von Zoledronsäure auf die Fertilität beim Menschen zu.
4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Nebenwirkungen wie Schwindel und Müdigkeit können einen Einfluss auf die Verkehrstüchtigkeit oder die Fähigkeit zum Bedienen von Maschinen haben, daher ist bei der Anwendung von Zoledronsäure zusammen mit Autofahren oder dem Bedienen von Maschinen Vorsicht geboten.
4.8 Nebenwirkungen
Zusammenfassung des Sicherheitsprofils
Innerhalb von drei Tagen nach Gabe von Zoledronsäure wurde häufig über eine Akute-Phase-Reaktion mit Symptomen wie Knochenschmerzen, Fieber, Müdigkeit, Arthralgie, Myalgie, Rigor und Arthritis mit darauf folgenden Gelenkschwellungen berichtet. Diese Symptome verschwinden üblicherweise innerhalb einiger Tage (siehe Beschreibung ausgewählter Nebenwirkungen).
Nachfolgend sind die wichtigen identifizierten Risiken mit Zoledronsäure in den zugelassenen Anwendungsgebieten aufgeführt: Nierenfunktionsstörung, Osteonekrose des Kieferknochens, Akute-Phase-Reaktion, Hypokalzämie, Vorhofflimmern, Anaphylaxie, interstitielle Lungenerkrankung. Die Häufigkeiten jedes dieser identifizierten Risiken sind in Tabelle 2 aufgeführt.
Tabellarische Auflistung von Nebenwirkungen
Die folgenden, in Tabelle 2 aufgeführten Nebenwirkungen sind in klinischen Studien und nach Markteinführung hauptsächlich bei chronischer Behandlung mit Zoledronsäure 4 mg aufgetreten.
Tabelle 2
Die Nebenwirkungen sind entsprechend ihrer Häufigkeit geordnet, die häufigste Nebenwirkung wird zuerst genannt.
Bei der Bewertung von Nebenwirkungen werden folgende Häufigkeiten zugrunde gelegt: sehr häufig (>1/10), häufig (>1/100, <1/10), gelegentlich (>1/1.000, <1/100), selten (>1/10.000, <1/1.000), sehr selten (<1/10.000), nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
|
Erkrankungen des Blutes und des Lymphsystems | |
|
Häufig: |
Anämie. |
|
Gelegentlich: |
Thrombozytopenie, Leukopenie. |
|
Selten: |
Panzytopenie. |
|
Erkrankungen des Immunsystems Gelegentlich: Selten: |
Überempfindlichkeitsreaktion. Angioneurotisches Ödem. |
|
Psychiatrische Erkrankungen Gelegentlich: |
Angstgefühle, Schlafstörungen. |
|
Selten: |
Verwirrung. |
|
Erkrankungen des Nervensystems Häufig: |
Kopfschmerzen. |
|
Gelegentlich: |
Schwindel, Parästhesien, Dysgeusie, Hypästhesie, Hyperästhesie, Tremor, Somnolenz. |
|
Sehr selten: |
Konvulsionen, Hypästhesie und Tetanie (durch Hypokalzämie) |
A ugenerkrankungen
|
Häufig: Gelegentlich: |
Konjunktivitis. Verschwommenes Sehen, Skleritis, Augenhöhlenentzündung. |
|
Selten: Sehr selten: |
Uveitis. Episkleritis. |
|
Herzerkrankungen Gelegentlich: |
Hypertonie, Hypotonie, Vorhofflimmern; Hypotonie, die zu Synkope oder Kreislaufkollaps führt. |
|
Selten: |
Bradykardie, Herzrhythmusstörungen (durch Hypokalzämie). |
Erkrankungen der Atemwege, des Brustraums und Mediastinums
Gelegentlich: Dyspnoe, Husten, Bronchokonstriktion
Selten:_Interstitielle Lungenerkrankung._
Erkrankungen des Gastrointestinaltrakts
Häufig: Übelkeit, Erbrechen, verminderter Appetit
|
Gelegentlich: |
Durchfall, Verstopfung, abdominale Schmerzen, Dyspepsie, Stomatitis, trockener Mund. |
Erkrankungen der Haut und des Unterhautzellgewebes
Gelegentlich: Pruritus, Ausschlag (einschließlich erythematöser und
makulärer Ausschlag), verstärktes Schwitzen.
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Häufig: Knochenschmerzen, Myalgie, Arthralgie,
|
Gelegentlich: Sehr selten |
generalisierte Schmerzen. Muskelspasmen, Osteonekrose des Kieferknochens1. Knochennekrose des äußeren Gehörgangs (Nebenwirkung der Arzneimittelklasse der Bisphosphonate). |
Erkrankungen der Nieren und Harnwege
Häufig: Nierenfunktionsstörungen.
Gelegentlich:_Akutes Nierenversagen, Hämaturie, Proteinurie
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Häufig: Fieber, grippeähnliche Symptome (einschließlich
|
Gelegentlich: |
Müdigkeit, Frösteln, Krankheitsgefühl, Flush). Asthenie, periphere Ödeme, Reaktionen an der Infusionsstelle (einschließlich Schmerz, Irritationen, Schwellung, Induration), Thoraxschmerzen, Gewichtszunahme, Anaphylaktische Reaktion/Schock, Urtikaria. |
|
Selten: |
Arthritis und Gelenkschwellung als Symptom einer Akute-Phase-Reaktion |
|
Untersuchungen Sehr häufig: Häufig: |
Hypophosphatämie. Erhöhung des Serum-Kreatinins und -Harnstoffs, Hypokalzämie. |
|
Gelegentlich: Selten: |
Hypomagnesiämie, Hypokaliämie. Hyperkaliämie, Hypernatriämie. |
1: Basierend auf klinischen Studien mit Beurteilung möglicher Fälle von Osteonekrose des Kieferknochens. Da diese Berichte durch unterschiedliche Faktoren beeinflusst werden, ist es nicht möglich, verlässlich einen ursächlichen Zusammenhang zur Exposition mit dem Arzneimittel herzustellen.
Beschreibung ausgewählter Nebenwirkungen
Nierenfunktionsstörunq
Zoledronsäure wurde mit Berichten über Nierenfunktionsstörunqen in Zusammenhang gebracht. In einer gemeinsamen Auswertung der Sicherheitsdaten aus den Zoledronsäure-Zulassungsstudien zur Prävention skelettbezogener Komplikationen bei Patienten mit fortgeschrittenen, auf das Skelett ausgedehnten Tumorerkrankungen war die Häufigkeit von Nierenfunktionsstörungen als unerwünschtes Ereignis mit einem Verdacht auf einen Zusammenhang mit Zoledronsäure (Nebenwirkung) wie folgt: Multiples Myelom (3,2%), Prostatakrebs (3,1%), Brustkrebs (4,3%), Lunge und andere solide Tumoren (3,2%). Faktoren, die die Möglichkeit einer Verschlechterung der Nierenfunktion erhöhen, sind: Dehydratation, vorbestehende Nierenfunktionsstörung, die mehrfache Anwendung von Zoledronsäure oder von anderen Bisphosphonaten sowie die gleichzeitige Anwendung nephrotoxischer Arzneimittel oder eine kürzere Infusionszeit als derzeit empfohlen. Über eine Verschlechterung der Nierenfunktion, ein Fortschreiten bis hin zum Nierenversagen und zur Dialyse wurde bei Patienten nach der ersten Dosis oder nach der einmaligen Gabe von 4 mg Zoledronsäure berichtet (siehe Abschnitt 4.4).
Osteonekrose des Kieferknochens
Über Osteonekrosen (vorwiegend im Kieferbereich) wurde in erster Linie bei Tumorpatienten berichtet, die mit Arzneimitteln, die die Knochenresorption hemmen, wie Zoledronsäure, behandelt wurden. Viele dieser Patienten hatten Anzeichen einer lokalen Infektion einschließlich Osteomyelitis. Die Mehrzahl der Berichte bezieht sich auf Tumorpatienten nach Zahnextraktion oder anderen dentalen Eingriffen. Es gibt zahlreiche, dokumentierte Risikofaktoren für Osteonekrosen der Kieferknochen einschließlich einer Tumordiagnose, verschiedener Begleittherapien (z.B. Chemo- oder Radiotherapie, Behandlung mit Kortikosteroiden) sowie gleichzeitig bestehender Erkrankungen (z.B. Anämien, Koagulopathien, Infektionen, vorbestehende Erkrankungen im Mundbereich). Obwohl keine Kausalität festgestellt wurde, wird empfohlen bei Patienten, die mit Zoledronsäure behandelt werden, vorsichtshalber dentale Eingriffe zu vermeiden, da es zu einer verzögerten Genesung kommen kann (siehe Abschnitt 4.4).
Vorhofflimmern
In einer randomisierten, doppelblind-kontrollierten Studie über 3 Jahre zur Bewertung der Sicherheit und Wirksamkeit von Zoledronsäure 5 mg einmal jährlich vs. Placebo zur Behandlung von postmenopausaler Osteoporose (PMO) betrug die Gesamthäufigkeit an Vorhofflimmern 2,5% (96 von 3.862) bzw. 1,9% (75 von 3.852) bei Patienten, die 5 mg Zoledronsäure bzw. Placebo erhielten. Die Häufigkeit von als schwerwiegende Ereignisse gemeldeten Fällen von Vorhofflimmern war bei Patienten, die Zoledronsäure 5 mg erhielten, 1,3% (51 von 3.862) im Vergleich zu 0,6% bei Patienten, die Placebo erhielten (22 von 3.852). Die in dieser Studie beobachtete Unausgewogenheit wurde in anderen Studien mit Zoledronsäure nicht beobachtet, einschließlich solcher Studien, die mit Zoledronsäure 4 mg alle 3-4 Wochen bei onkologischen Patienten durchgeführt wurden. Der Mechanismus hinter der vermehrten Häufigkeit an Vorhofflimmern in dieser einzelnen Studie ist unbekannt.
Akute-Phase-Reaktion
Diese Nebenwirkung beinhaltet eine Reihe von Symptomen wie Fieber,
Myalgie, Kopfschmerzen, Schmerzen in den Extremitäten, Übelkeit, Erbrechen, Diarrhö, Arthralgie und Arthritis mit darauf folgenden Gelenkschwellungen. Diese treten innerhalb von <3 Tagen nach der Infusion von Zoledronsäure auf. Die Reaktion wird auch als „grippeähnlich" oder als „Postinfusions-Symptom" bezeichnet.
Atypische Femurfrakturen
Nach Markteinführung wurden die folgenden Nebenwirkungen berichtet (Häufigkeit: selten): Atypische subtrochantäre und diaphysäre Femurfrakturen (unerwünschte Wirkung der Substanzklasse der Bisphosphonate).
Nebenwirkungen in Zusammenhang mit Hypokalzämie Hypokalzämie ist ein wesentliches identifiziertes Risiko für Zoledronsäure in den zugelassenen Anwendungsgebieten. Basierend auf der Bewertung von Fällen aus klinischen Studien und nach Markteinführung gibt es ausreichend Hinweise, die einen Zusammenhang zwischen einer Behandlung mit Zoledronsäure, dem berichteten Ereignis Hypokalzämie und der daraus folgenden Entwicklung von Herzrhythmusstörungen unterstützen. Darüber hinaus gibt es Anzeichen für einen Zusammenhang zwischen Hypokalzämie und daraus folgenden neurologischen Ereignissen, die für diese Fälle berichtet wurden, einschließlich Konvulsionen, Hypästhesie und Tetanie (siehe Abschnitt 4.4).
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem
Bundesinstitut für Arzneimittel und Medizinprodukte Abt. Pharmakovigilanz Kurt-Georg-Kiesinger-Allee 3 D-53175 Bonn Website: www.bfarm.de
anzuzeigen.
4.9 Überdosierung
Klinische Erfahrungen über akute Überdosierung mit Zoledronsäure sind begrenzt. Die versehentliche Anwendung von Dosen bis zu 48 mg Zoledronsäure wurde berichtet. Patienten, die eine höhere als die empfohlene Dosierung (siehe Abschnitt 4.2) erhalten haben, müssen sorgfältig überwacht werden, da eine eingeschränkte Nierenfunktion (einschließlich Nierenversagen) und Veränderungen der Elektrolyte im Serum (einschließlich Calcium, Phosphor und Magnesium) beobachtet wurden. Im Falle einer klinisch relevanten Hypokalzämie müssen Calciumgluconat-Infusionen, wie klinisch angezeigt, verabreicht werden.
5. Pharmakologische Eigenschaften
5.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Mittel zur Behandlung von Knochenerkrankungen, Bisphosphonate ATC-Code: M05BA08
Zoledronsäure gehört zur Gruppe der Bisphosphonate und wirkt primär am Knochen. Sie ist ein Inhibitor der osteoklastischen Knochenresorption.
Die selektive Wirkung von Bisphosphonaten auf das Knochengewebe ist durch ihre hohe Affinität zum Knochenmineral bedingt. Der genaue molekulare Wirkungsmechanismus, der zur Hemmung der Osteoklastenaktivität führt, ist bisher jedoch nicht bekannt. In Langzeituntersuchungen am Tier hemmte Zoledronsäure die Knochenresorption, ohne die Neubildung, die Mineralisation oder die mechanischen Eigenschaften des Knochens nachteilig zu beeinflussen.
Zusätzlich zu ihrer Eigenschaft als potenter Inhibitor der Knochenresorption besitzt Zoledronsäure verschiedene Anti-Tumor-Eigenschaften, die zur Gesamtwirkung der Substanz bei der Behandlung von metastatischen Knochenveränderungen beitragen könnten. Die folgenden Eigenschaften wurden in präklinischen Studien nachgewiesen:
• In vivo: Hemmung der durch Osteoklasten verursachten Knochenresorption, wodurch das Mikro-Milieu des Knochenmarks verändert und dadurch weniger anfällig für das Wachstum von Tumorzellen wird. Außerdem: Anti-Angiogenese-Aktivität und analgetischer Effekt.
• In vitro: Hemmung der Osteoblastenproliferation; direkte zytostatische und pro-apoptotische Aktivität auf Tumorzellen; synergistischer zytostatischer Effekt mit anderen anti-tumorösen Arzneimitteln und Anti-Adhäsions/Invasions-Wirkung.
Ergebnisse klinischer Studien bei der Prävention skelettbezogener Komplikationen bei Patienten mit fortgeschrittenen, auf das Skelett ausgedehnten Tumorerkrankungen
In der ersten randomisierten, doppelblinden, placebokontrollierten Studie wurden Zoledronsäure 4 mg und Placebo zur Prävention von Skelettkomplikationen („Skeletal Related Events" = SREs) bei Patienten mit Prostatakarzinom verglichen. Zoledronsäure 4 mg reduzierte signifikant den Anteil der Patienten, die mindestens eine SRE erlitten, verzögerte die Zeit (median) bis zum Auftreten der ersten SRE um mehr als 5 Monate und verringerte die skelettale Morbiditätsrate (Anzahl der SREs pro Patient und Jahr). Eine Multiple-Event-Analyse zeigte in der Zoledronsäure 4 mg-Gruppe eine 36%ige Risikoreduktion für das Auftreten von SREs im Vergleich zu Placebo. Unter Zoledronsäure 4 mg berichteten die Patienten über eine geringere Schmerzzunahme als unter Placebo. Dieser Unterschied war nach 3, 9, 21 und 24 Monaten signifikant. Weniger Zoledronsäure 4 mg-Patienten erlitten pathologische Frakturen. Die Behandlungseffekte waren bei Patienten mit blastischen Läsionen weniger ausgeprägt. Die Ergebnisse zur Wirksamkeit sind in Tabelle 3 zusammengefasst.
In einer zweiten Studie zu anderen soliden Tumoren als Mamma- oder Prostatakarzinomen reduzierte Zoledronsäure 4 mg signifikant den Anteil der Patienten mit einer SRE, verlängerte im Median die Zeit bis zum ersten Auftreten einer SRE um mehr als 2 Monate und verringerte die skelettale Morbiditätsrate. Eine Multiple-Event-Analyse zeigte in der Zoledronsäure 4 mgGruppe eine 30,7%ige Risikoreduktion für SREs im Vergleich zu Placebo. Die Ergebnisse zur Wirksamkeit sind in Tabelle 4 zusammengefasst.
Tabelle 3: Ergebnisse zur Wirksamkeit (Patienten mit Prostatakarzinom unter
|
hormoneller T |
nerapie) | |||||
|
SRE (+TIH) |
Frakturen* |
Radiotherapie am Knochen | ||||
|
Zoledronsäure 4 mg |
Placebo |
Zoledronsäure 4 mg |
Placebo |
Zoledronsäure 4 mg |
Placebo | |
|
Anzahl(N) |
214 |
208 |
214 |
208 |
214 |
208 |
|
Anteil Patienten mit SREs (%) |
38 |
49 |
17 |
25 |
26 |
33 |
|
p-Wert |
0,028 |
0,052 |
0,119 | |||
|
Zeit bis zum Auftreten der ersten SRE in Tagen (median) |
488 |
321 |
NE |
NE |
NE |
640 |
|
p-Wert |
0,009 |
0,020 |
0,055 | |||
|
Skelettale Morbiditätsrate |
0,77 |
1,47 |
0,20 |
0,45 |
0,42 |
0,89 |
|
p-Wert |
0,005 |
0,023 |
0,060 | |||
|
Riskoreduktion gemäß Multiple-Event-Analyse** (%) |
36 |
NZ |
NZ |
NZ |
NZ | |
|
p-Wert |
0,002 |
NZ |
NZ | |||
* vertebrale und nicht-vertebrale Frakturen
** Alle skelettalen Ereignisse, sowohl gesamte Anzahl als auch Zeit bis zum Erreichen jedes Ereignisses während der Studie NE Nicht erreicht
NZ Nicht zutreffend
|
SRE (+TIH) |
Frakturen* |
Radiotherapie am Knochen | ||||
|
Zoledronsäure 4 mg |
Placebo |
Zoledronsäure 4 mg |
Placebo |
Zoledronsäure 4 mg |
Placebo | |
|
Anzahl(N) |
257 |
250 |
257 |
250 |
257 |
250 |
|
Anteil Patienten mit SREs(%) |
39 |
48 |
16 |
22 |
29 |
34 |
|
p-Wert |
0,039 |
0,064 |
0,173 | |||
|
Zeit bis zum Auftreten der ersten SRE in Tagen (median) |
236 |
155 |
NE |
NE |
424 |
307 |
|
p-Wert |
0,009 |
0,020 |
0,079 | |||
|
Skelettale Morbiditätsrate |
1,74 |
2,71 |
0,39 |
0,63 |
1,24 |
1,89 |
|
p-Wert |
0,012 |
0,066 |
0,099 | |||
|
Riskoreduktion gemäß Multiple-Event-Analyse** (%) |
30,7 |
NZ |
NZ |
NZ |
NZ | |
|
p-Wert |
0,003 |
NZ |
NZ | |||
Tabelle 4: Ergebnisse zur Wirksamkeit (solide Tumoren außer Mammakarzinom und Prostatakarzinom)_
* vertebrale und nicht-vertebrale Frakturen
** Alle skelettalen Ereignisse, sowohl gesamte Anzahl als auch Zeit bis zum Erreichen jedes Ereignisses während der Studie NE Nicht erreicht
NZ Nicht zutreffend
In einer dritten doppelblinden, randomisierten Phase-III-Studie wurde die Anwendung von Zoledronsäure 4 mg oder 90 mg Pamidronsäure jeweils alle 3 bis 4 Wochen bei Patienten mit Multiplem Myelom oder Mammakarzinom und mindestens einer Knochenläsion verglichen. Die Ergebnisse zeigten, dass Zoledronsäure 4 mg in der Prävention skelettbezogener Ereignisse eine vergleichbare Wirksamkeit aufweist wie 90 mg Pamidronsäure. Die Multiple-Event-Analyse zeigte in der Zoledronsäure-4 mg-Gruppe eine signifikante 16%ige Risikoreduktion im Vergleich zu Patienten, die Pamidronsäure erhalten hatten. Die Ergebnisse zur Wirksamkeit sind in Tabelle 5 zusammengefasst.
Tabelle 5: Ergebnisse zur Wirksamkeit (Patienten mit Mammakarzinom oder Multiplem Myelom)
|
SRE (+TIH) |
Frakturen* |
Radiotherapie am Knochen | ||||
|
Zoledronsäure 4 mg |
Pam 90 mg |
Zoledronsäure 4 mg |
Pam 90 mg |
Zoledronsäure 4 mg |
Pam 90 mg | |
|
Anzahl(N) |
561 |
555 |
561 |
555 |
561 |
555 |
|
Anteil Patienten mit SREs (%) |
48 |
52 |
37 |
39 |
19 |
24 |
|
p-Wert |
0,198 |
0,653 |
0,037 | |||
|
Zeit bis zum Auftreten der ersten SRE in Tagen (median) |
376 |
356 |
NE |
714 |
NE |
NE |
|
p-Wert |
0,151 |
0,672 |
0,026 | |||
|
Skelettale Morbiditätsrate |
1,04 |
1,39 |
0,53 |
0,60 |
0,47 |
0,71 |
|
p-Wert |
0,084 |
0,614 |
0,015 | |||
|
Riskoreduktion gemäß Multiple-Event-Analyse** (%) events** |
16 |
NZ |
NZ |
NZ |
NZ | |
|
p-Wert |
0,030 |
NZ |
NZ | |||
* vertebrale und nicht-vertebrale Frakturen
** Alle skelettalen Ereignisse, sowohl gesamte Anzahl als auch Zeit bis zum Erreichen jedes Ereignisses während der Studie NE Nicht erreicht
NZ Nicht zutreffend
Zoledronsäure 4 mg wurde auch in einer doppelblinden, randomisierten, placebokontrollierten Studie an 228 Patienten mit dokumentierten Knochenmetastasen nach Mammatumor untersucht, um die Wirkung von 4 mg Zoledronsäure auf die Skelettkomplikationen (SRE) zu bewerten, berechnet als Gesamtzahl der SRE-Ereignisse (mit Ausnahme von Hyperkalzämie und an vorhergehende Frakturen angepasst), geteilt durch den gesamten Risikozeitraum. Die Patienten erhielten für ein Jahr alle vier Wochen entweder 4 mg Zoledronsäure oder Placebo. Die Patienten wurden gleichmäßig zwischen den Zoledronsäure-behandelten und Placebo-Gruppen aufgeteilt.
Die SRE-Rate (Ereignisse/Personenjahr) beträgt für Zoledronsäure 0,628 und für Placebo 1,096. Das Verhältnis von Patienten mit zumindest einer SRE (mit Ausnahme von Hyperkalzämie) betrug 29,8% in der mit Zoledronsäure behandelten Gruppe vs. 49,6% in der Placebo-Gruppe (p=0,003). In dem mit Zoledronsäure behandelten Arm wurde am Ende der Studie die mediane Zeit bis zum Auftreten des ersten SRE nicht erreicht und war im Vergleich zu Placebo signifikant verzögert (p=0,007). In einer Analyse von Mehrfachereignissen verringerte Zoledronsäure 4 mg das Risiko für SREs um 41% (Risiko-Verhältnis 0,59, p=0,019) im Vergleich zu Placebo.
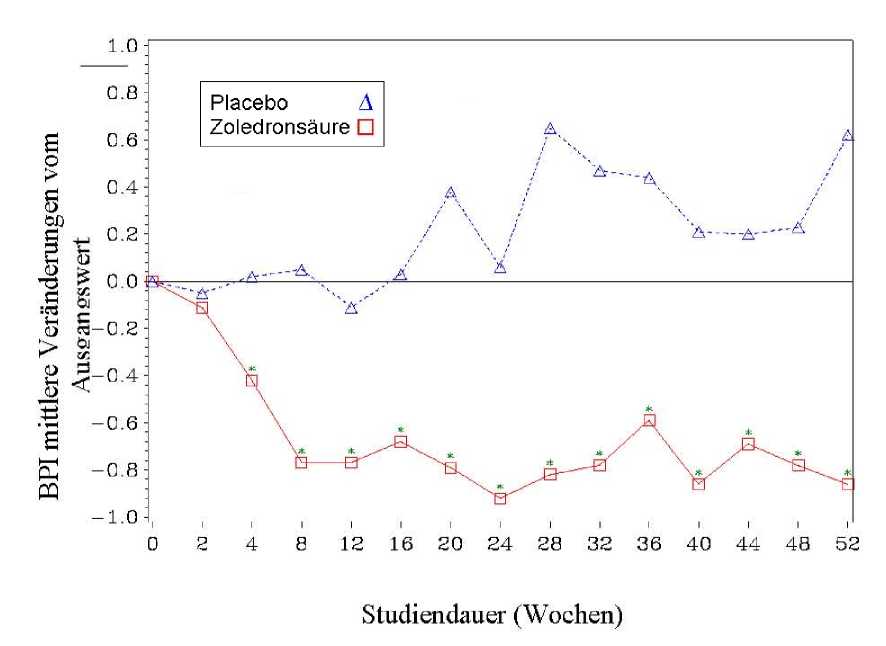
In der mit Zoledronsäure behandelten Gruppe wurde eine statistisch signifikante Verbesserung des Schmerz-Scores (unter Verwendung des Brief Pain Inventory, BPI) nach 4 Wochen gesehen und zu jedem nachfolgenden Zeitpunkt während der Studie, wenn mit Placebo verglichen wurde (Abbildung 1). Für Zoledronsäure lag der Schmerz-Score durchweg unterhalb des Ausgangswertes und die Schmerzverminderung wurde tendenziell von einer Reduktion des Schmerzmittel-Scores begleitet.
Abbildung 1: Mittlere Veränderungen der BPI-Scores vom Ausgangswert. Statistisch signifikante Unterschiede sind gekennzeichnet (*p<0,05) für den Vergleich der Behandlung (4 mg Zoledronsäure vs. Placebo)
Ergebnisse klinischer Studien in der Behandlung der TIH Klinische Studien bei tumorinduzierter Hyperkalzämie (TIH) zeigten, dass die Wirkung von Zoledronsäure durch eine Abnahme des Serum-Calciums und der Calcium-Ausscheidung im Urin gekennzeichnet ist. In Phase-I-Dosisfindungsstudien an Patienten mit leichter bis mittelschwerer tumorinduzierter Hyperkalzämie (TIH) lagen die untersuchten, wirksamen Dosierungen im Bereich von ca. 1,2-2,5 mg.
Zum Nachweis der Wirksamkeit von 4 mg Zoledronsäure im Vergleich zu 90 mg Pamidronsäure wurden die Ergebnisse von zwei pivotalen, multizentrischen
Studien an Patienten mit TIH in einer vorher geplanten Analyse kombiniert. Es erfolgte eine schnellere Normalisierung des korrigierten Serum-Calciums am Tag 4 mit 8 mg Zoledronsäure und am Tag 7 mit 4 mg und 8 mg Zoledronsäure. Die folgenden Ansprechraten wurden beobachtet:
Tabelle 6: Komplette Ansprechrate pro Tag in den kombinierten TIH-Studien
|
Tag 4 |
Tag 7 |
Tag 10 | |
|
Zoledronsäure 4 mg (N=86) |
45,3% (p=0,104) |
82,6% (p=0,005)* |
88,4% (p=0,002)* |
|
Zoledronsäure 8 mg (N=90) |
55,6% (p=0,021)* |
83,3% (p=0,010)* |
86,7% (p=0,015)* |
|
Pamidronsäure 90 mg (N=99) |
33,3% |
63,6% |
69,7% |
|
*p-Werte im Vergleich zu Pamidronat | |||
Im Median betrug die Zeit bis zum Erreichen normokalzämischer Werte 4 Tage. Die mediane Dauer bis zum Rezidiv (Wiederanstieg der Albumin-korrigierten Serum-Calcium-Spiegel auf >2,9 mmol/l) betrug 30-40 Tage bei Patienten, die mit Zoledronsäure behandelt wurden, gegenüber 17 Tagen bei denjenigen, die mit 90 mg Pamidronsäure behandelt wurden (p-Werte: 0,001 für 4 mg und
0. 007 für 8 mg Zoledronsäure). Zwischen beiden Zoledronsäure-Dosierungen gab es keine statistisch signifikanten Unterschiede.
In klinischen Studien erhielten 69 Patienten, die gegenüber der ersten Behandlung (4 mg oder 8 mg Zoledronsäure oder 90 mg Pamidronat) refraktär waren, eine Wiederbehandlung mit 8 mg Zoledronsäure. Die Ansprechrate betrug bei diesen Patienten ca. 52%. Da diese Patienten ausschließlich mit der 8 mg-Dosis wiederbehandelt wurden, sind keine Daten verfügbar, die einen Vergleich mit der 4 mg-Zoledronsäure-Dosis erlauben würden.
In klinischen Studien an Patienten mit tumorinduzierter Hyperkalzämie (TIH) war das gesamte Sicherheitsprofil zwischen allen drei Behandlungsgruppen (4 mg und 8 mg Zoledronsäure und 90 mg Pamidronsäure) hinsichtlich Art und Schweregrad vergleichbar.
Kinder und Jugendliche
Ergebnisse der klinischen Studien zur Behandlung der schweren Osteogenesis imperfecta bei Kindern und Jugendlichen im Alter von 1 bis 17 Jahren Die Wirkung von intravenöser Zoledronsäure zur Behandlung von Kindern und Jugendlichen (Alter 1 bis 17 Jahre) mit schwerer Osteogenesis imperfecta (Typ
1, III und IV) wurde im Vergleich zu intravenösem Pamidronat in einer internationalen, multizentrischen, randomisierten, offenen Studie mit 74 bzw. 76 Patienten in der jeweiligen Behandlungsgruppe untersucht. Der Behandlungszeitraum in der Studie betrug 12 Monate. Diesem ging eine 4- bis 9-wöchige Screening-Phase voraus, während der über mindestens 2 Wochen Vitamin D und Calcium eingenommen wurden. In klinischen Studien erhielten Patienten im Alter von 1 bis <3 Jahren 0,025 mg/kg Zoledronsäure (bis zu einer maximalen Einzeldosis von 0,35 mg) alle 3 Monate. Patienten im Alter von 3 bis 17 Jahren wurden 0,05 mg/kg Zoledronsäure (bis zu einer maximalen Einzeldosis von 0,83 mg) alle 3 Monate verabreicht. Es wurde eine Extensionsstudie durchgeführt, um die allgemeine und renale Langzeitsicherheit einer einmal bzw. zweimal jährlichen Gabe von Zoledronsäure während der 12-monatigen Extension bei Kindern, die die Behandlung in der 1-Jahres-Hauptstudie mit Zoledronsäure oder Pamidronat abgeschlossen hatten, zu untersuchen.
Der primäre Endpunkt der Studie war die prozentuale Änderung der Knochendichte (BMD) der Lendenwirbelsäule nach 12 Monaten Behandlung. Die geschätzte Wirkung auf die BMD war ähnlich, aber das Studiendesign war nicht ausreichend robust um eine „Nicht-Unterlegenheit" der Wirksamkeit von Zoledronsäure nachzuweisen. Vor allem gab es keine eindeutigen Beweise einer Wirksamkeit auf die Frakturhäufigkeit und bei Schmerzen. An den langen Knochen der unteren Extremitäten wurden Frakturen als unerwünschte Ereignissen bei ca. 24% (Femur) und 14% (Tibia) der mit Zoledronsäure und bei 12% bzw. 5% der mit Pamidronat behandelten Patienten mit schwerer Osteogenesis imperfecta, unabhängig von der Grunderkrankung oder eines Kausalzusammenhangs, beobachtet. Die Gesamthäufigkeit für Frakturen war bei Zoledronsäure- bzw. bei Pamidronatpatienten vergleichbar, nämlich 43% (32/74) bzw. 41% (31/76). Die Interpretation des Frakturrisikos wird durch die Tatsache erschwert, dass Frakturen, aufgrund des Fortschreitens der Grunderkrankung, häufige Ereignisse bei Patienten mit schwerer Osteogenesis imperfecta sind.
Die Art der Nebenwirkungen, die in dieser Population beobachtet wurden, war ähnlich wie bei Erwachsenen mit fortgeschrittenen Tumorerkrankungen unter Beteiligung der Knochen (siehe Abschnitt 4.8). Die Nebenwirkungen sind in Tabelle 7 nach ihrer Häufigkeit geordnet aufgeführt. Bei der Bewertung von Nebenwirkungen werden folgende Häufigkeiten zugrunde gelegt: sehr häufig (>1/10), häufig (>1/100, <1/10), gelegentlich (>1/1.000, <1/100), selten (>1/10.000, <1/1.000), sehr selten (<1/10.000), nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
Tabelle 7: Nebenwirkungen bei Kindern und Jugendlichen mit schwerer Osteogenesis imperfecta1
|
Erkrankungen des Nervensystems Häufig: |
Kopfschmerzen. |
|
Herzerkrankungen | |
|
Häufig: |
Tachykardie. |
|
Erkrankungen der Atemwege, des Brustraums und Mediastinums | |
|
Häufig: |
Nasopharyngitis. |
Erkrankungen des Gastrointestinaltrakts
Sehr häufig: Übelkeit, Erbrechen.
Häufig: Abdominale Schmerzen.
|
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen | |
|
Häufig: |
Schmerzen in den Extremitäten, Arthralgie, muskuloskelettale Schmerzen. |
|
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort | |
|
Sehr häufig: |
Fieber, Müdigkeit. |
|
Häufig: |
Akute-Phase-Reaktion, Schmerzen. |
|
Untersuchungen Sehr häufig: |
Hypokalzämie. |
|
Häufig: |
Hypophosphatämie. |
1 Unerwünschte Ereignisse mit einer Häufigkeit von <5% wurden medizinisch bewertet und es wurde gezeigt, dass diese Fälle mit dem gut bekannten Sicherheitsprofil von Zoledronsäure übereinstimmen (siehe Abschnitt 4.8).
Bei Kindern und Jugendlichen mit schwerer Osteogenesis imperfecta scheint Zoledronsäure im Vergleich zu Pamidronat mit einem höheren Risiko für AkutePhase-Reaktionen, Hypokalzämie und ungeklärte Tachykardie verbunden zu sein. Dieser Unterschied verringerte sich nach den weiteren Infusionen.
Die Europäische Arzneimittel-Agentur hat Zoledronsäure von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in allen pädiatrischen Gruppierungen für die Behandlung der tumorinduzierten Hyperkalzämie und zur Prävention skelettbezogener Komplikationen bei Patienten mit fortgeschrittenen, auf das Skelett ausgedehnten Tumorerkrankungen freigestellt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen).
5.2 Pharmakokinetische Eigenschaften
Eine einmalige oder mehrfache 5- und 15-minütige Infusion von 2, 4, 8 und 16 mg Zoledronsäure bei 64 Patienten mit Knochenmetastasen ergab die folgenden dosisunabhängigen pharmakokinetischen Daten:
Nach Start der Zoledronsäure-Infusion erhöht sich die Plasmakonzentration von Zoledronsäure schnell, wobei die Plasmaspitzenkonzentration am Ende der Infusionszeit erreicht wird. Es folgt ein schneller Rückgang auf <10% der Plasmaspitzenkonzentration nach 4 Stunden und auf <1% nach 24 Stunden, gefolgt von einem längeren Zeitraum mit sehr niedrigen Konzentrationen, die nicht über 0,1% der Plasmaspitzenkonzentration hinausgehen, bevor am Tag 28 die zweite Infusion von Zoledronsäure erfolgt.
Die Ausscheidung von intravenös verabreichter Zoledronsäure verläuft triphasisch: Eine schnelle, biphasische Elimination aus der systemischen Zirkulation mit Halbwertszeiten von t%a 0,24 und t%ß 1,87 Stunden, gefolgt von einer lang andauernden Eliminationsphase mit einer terminalen Eliminationshalbwertszeit von t%Y 146 Stunden. Auch nach Mehrfachgabe (alle 28 Tage) kommt es nicht zur Akkumulation von Zoledronsäure im Plasma. Zoledronsäure wird nicht metabolisiert, sondern unverändert über die Nieren ausgeschieden. Innerhalb der ersten 24 Stunden werden 39 ± 16% der verabreichten Dosis im Urin wiedergefunden, während die Restmenge prinzipiell am Knochengewebe gebunden ist. Aus dem Knochengewebe wird Zoledronsäure sehr langsam zurück in den systemischen Kreislauf abgegeben und über die Nieren ausgeschieden. Die Gesamtkörper-Clearance beträgt unabhängig von der Dosierung 5,04 ± 2,5 l/h und wird durch Geschlecht, Alter, Rasse und Körpergewicht nicht beeinflusst. Eine Erhöhung der Infusionszeit von 5 auf 15 Minuten führte zu einer Abnahme der Zoledronsäure-Konzentration am Ende der Infusion um 30%, hatte aber keinen Einfluss auf die AUC im Plasmakonzentration-Zeit-Diagramm.
Wie bei anderen Bisphosphonaten ist die Variabilität der pharmakokinetischen Parameter von Zoledronsäure zwischen den Patienten hoch.
Pharmakokinetische Daten zu Zoledronsäure bei Patienten mit Hyperkalzämie sowie bei Patienten mit Leberinsuffizienz liegen nicht vor. Zoledronsäure hemmt in vitro keine humanen P450-Enzyme und wird nicht metabolisiert. In Tierstudien wurden <3% der verabreichten Dosis in den Fäzes wiedergefunden. Dies deutet darauf hin, dass die Leberfunktion keine relevante Rolle für die Pharmakokinetik von Zoledronsäure spielt.
Die renale Clearance von Zoledronsäure korreliert mit der Kreatinin-Clearance. Die renale Clearance entspricht 75 ± 33% der Kreatinin-Clearance, die bei den 64 untersuchten Tumorpatienten im Mittel bei 84 ± 29 ml/min (von 22 bis 143 ml/min) lag. Eine Populationsanalyse zeigte für Patienten mit einer Kreatinin-Clearance von 20 ml/min (schwere Niereninsuffizienz) bzw. 50 ml/min (mittelschwere Niereninsuffizienz), dass die voraussagbare Clearance von Zoledronsäure 37% bzw. 72% derjenigen eines Patienten mit einer Kreatinin-Clearance von 84 ml/min betragen würde. Für Patienten mit schweren Nierenfunktionsstörungen (Kreatinin-Clearance <30 ml/min) liegen nur wenige pharmakokinetische Daten vor.
In einer In-vitro-Studie zeigte Zoledronsäure in einem Konzentrationsbereich von 30 ng/ml bis 5.000 ng/ml eine geringe Affinität zu menschlichen Blutzellen, mit einem mittleren Blut/Plasmakonzentrationsverhältnis von 0,59. Die Plasmaproteinbindung ist gering, wobei der ungebundene Anteil zwischen 60% bei 2 ng/ml und 77% bei 2.000 ng/ml Zoledronsäure liegt.
Spezielle Patientengruppen
Pädiatrische Patienten
Begrenzte pharmakokinetische Daten bei Kindern und Jugendlichen mit schwerer Osteogenesis imperfecta legen nahe, dass die Pharmakokinetik von Zoledronsäure bei Kindern und Jugendlichen im Alter von 3 bis 17 Jahren bei ähnlicher mg/kg-Dosis vergleichbar mit derjenigen von Erwachsenen ist. Alter, Körpergewicht, Geschlecht und Kreatinin-Clearance haben offensichtlich keinen Effekt auf die systemische Exposition von Zoledronsäure.
5.3 Präklinische Daten zur Sicherheit
Akute Toxizität
Die höchste nicht letal wirkende intravenöse Einzeldosis betrug bei Mäusen 10 mg/kg Körpergewicht und bei Ratten 0,6 mg/kg Körpergewicht.
Subchronische und chronische Toxizität
Bis zu einer täglichen Dosis von 0,02 mg/kg Körpergewicht über 4 Wochen wurde Zoledronsäure bei subkutaner Gabe von Ratten und bei intravenöser Gabe von Hunden gut vertragen. Die subkutane Gabe von 0,001 mg/kg/Tag an Ratten und die intravenöse Gabe von 0,005 mg/kg einmal alle 2-3 Tage an Hunden über einen Zeitraum von bis zu 52 Wochen wurde ebenfalls gut vertragen.
In den Studien mit wiederholter Gabe war bei nahezu allen Dosierungen der häufigste Befund eine Zunahme der primären Spongiosa in der Metaphyse langer Knochen bei wachsenden Tieren. Dieser Befund spiegelt die pharmakologische, antiresorptive Wirkung der Substanz wider.
In den Langzeitstudien mit wiederholter parenteraler Gabe am Tier zeigte sich, dass der Sicherheitsabstand hinsichtlich renaler Effekte klein ist. Die kumulativen NOAELs („no observed adverse effect levels") in den Studien mit Einzelgabe (1,6 mg/kg) und Mehrfachgabe bis zu einem Monat (0,06-0,6 mg/kg/Tag) ergaben jedoch keine Hinweise auf renale Effekte bei Dosierungen, die der höchsten vorgesehenen therapeutischen Humandosis entsprachen oder diese übertrafen. Wiederholte Gaben über einen längeren Zeitraum bei Dosierungen rund um die höchste vorgesehene therapeutische Humandosis von Zoledronsäure führten zu toxikologischen Wirkungen in anderen Organen einschließlich Gastrointestinaltrakt, Leber, Milz, Lunge und an der intravenösen Injektionsstelle.
Reproduktionstoxikologie
Zoledronsäure war teratogen bei der Ratte bei subkutanen Dosen von >0,2 mg/kg. Obwohl beim Kaninchen keine Teratogenität oder Fetotoxizität beobachtet wurden, wurde maternale Toxizität gefunden. Bei Ratten wurde bei der niedrigsten untersuchten Dosis (0,01 mg/kg Körpergewicht) erschwerte Geburt (Dystokie) beobachtet.
Mutagenität und Kanzerogenität
In den durchgeführten Mutagenitätstests erwies sich Zoledronsäure als nicht mutagen. Studien zur Kanzerogenität lieferten keine Hinweise auf ein kanzerogenes Potenzial.
6. Pharmazeutische Angaben
6.1 Liste der sonstigen Bestandteile
Mannitol (Ph.Eur.)
Natriumcitrat (Ph.Eur.)
Wasser für Injektionszwecke
6.2 Inkompatibilitäten
Dieses Arzneimittel darf nicht mit calciumhaltigen Lösungen in Kontakt gebracht werden und darf nicht mit anderen Arzneimitteln gemischt oder intravenös über dieselbe Infusionslinie verabreicht werden.
6.3 Dauer der Haltbarkeit
Ungeöffnete Flasche: 2 Jahre.
Nach dem ersten Öffnen:
Die chemische und physikalische Stabilität nach Anbruch wurde bei 25°C und 2°C - 8°C für 24 Stunden nachgewiesen. Aus mikrobiologischer Sicht sollte die verdünnte Infusionslösung sofort verwendet werden. Falls sie nicht sofort verwendet wird, ist der Anwender für die Dauer und Bedingungen der Aufbewahrung nach Zubereitung verantwortlich, die normalerweise 24 Stunden bei 2°C - 8°C nicht überschreiten sollte. Vor der Anwendung ist die gekühlte Lösung dann wieder auf Raumtemperatur zu bringen.
6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Für dieses Arzneimittel sind keine besonderen Lagerungsbedingungen erforderlich.
Aufbewahrungsbedingungen der Infusionslösung nach dem ersten Öffnen des Arzneimittels siehe Abschnitt 6.3
6.5 Art und Inhalt des Behältnisses
Durchsichtige Typ 1, innen mit Silicondioxid beschichtete, GlasDurchstechflaschen oder durchsichtige Typ 1 BorosilikatglasDurchstechflaschen oder durchsichtige Kunststoff-Durchstechflaschen aus Cyclo-Olefin-Copolymere (COC) mit einem Typ I Brombutyl-Gummistopfen und Aluminium-Polypropylen-Flip-off-Sicherheitskappe.
Zoledronsäure AL 4 mg/100 ml Infusionslösung ist in Packungen mit 1 und 4 Durchstechflaschen erhältlich
6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung
Weitere Informationen zur Handhabung von Zoledronsäure einschließlich einer Anweisung zur Herstellung verringerter Dosierungen unter Verwendung der Zoledronsäure-Fertiglösung befinden sich in Abschnitt 4.2.
Die Infusion ist unter aseptischen Bedingungen herzustellen. Nur zur einmaligen Anwendung bestimmt.
Es dürfen nur klare Lösungen ohne Partikel und Verfärbungen verwendet werden.
Das medizinische Fachpersonal ist anzuweisen, nicht verwendete Zoledronsäure nicht über das lokale Abwassersystem zu entsorgen.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu entsorgen.
7. Inhaber der Zulassung
ALIUD PHARMA® GmbH Gottlieb-Daimler-Str. 19 D-89150 Laichingen Telefon: 07333 9651-0 Telefax: 07333 9651-6004 info@aliud.de
8. Zulassungsnummer
88751.00.00
9. Datum der Erteilung der Zulassung/Verlängerung der Zulassung
18.09.2013
10. Stand der Information
Juli 2016
11. Verkaufsabgrenzung
Verschreibungspflichtig