Atrovent 500 Μg/2 Ml Fertiginhalat
F achinform ation
Boehringer n| \v Ingelheim
1. BEZEICHNUNG DES ARZNEIMITTELS
Atrovent® 500 ^g/2 ml Fertiginhalat Lösung für einen Vernebler
2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
Wirkstoff: Ipratropiumbromid 1 Eindosisbehälter zu 2 ml Lösung enthält:
522 ^g Ipratropiumbromid 1 H2O (entspricht 500 ^g Ipratropiumbromid) Vollständige Auflistung der sonstigen Bestandteile: siehe Abschnitt 6.1.
3. DARREICHUNGSFORM
Lösung für einen Vernebler
4. KLINISCHE ANGABEN
4.1 Anwendungsgebiete
Zur Verhütung und Behandlung von Atemnot bei
• chronisch obstruktiver Lungenerkrankung (COPD)
• leichtem bis mittelschwerem Asthma bronchiale im Erwachsenen- und Kindesalter, als Ergänzung zu ß2-Mimetika im akuten Asthmaanfall.
4.2 Dosierung und Art der Anwendung
Zur Inhalation mit einem Vernebler Dosierung
Die Dosierung muss dem Einzelfall angepasst werden.
Die folgenden Dosierungen werden empfohlen:
Zur Akutbehandlung:
Erwachsene und Jugendliche > 12 Jahre :
Die inhalative Einzeldosis liegt bei 0,5 mg Ipratropiumbromid, entsprechend 1 Eindosisbehälter; wiederholte Gaben können bis zur Besserung der Atemnot verabreicht werden, der zeitliche Abstand der einzelnen Inhalationen muss vom Arzt bestimmt werden.
Zur Dauerbehandlung:
Erwachsene und Jugendliche > 12 Jahre :
1 Eindosisbehälter 3 - 4-mal täglich.
Hinweis
Eine Tagesdosis von mehr als 2 mg Ipratropiumbromid (4 Eindosisbehälter) ist vom Arzt regelmäßig zu überprüfen.
Wichtiger Hinweis
Der Patient sollte während der Behandlung medizinisch überwacht werden.
Kommt es trotz der verordneten Therapie zu keiner befriedigenden Besserung oder gar zu einer Verschlechterung des Leidens, ist ärztliche Beratung erforderlich, um die Therapie ggf. unter Hinzuziehung anderer Arzneimittel (Kortikoide, ß2-Sympathikomimetika, Theophyllin) neu festzulegen. Der Patient muss angewiesen werden bei akuter oder sich rasch verschlimmernder Atemnot unverzüglich ärztliche Hilfe in Anspruch zu nehmen.
Art der Anwendung
Diese Lösung ist gebrauchsfertig, d. h. eine Verdünnung ist nicht erforderlich. Die Lösung in den Eindosisbehältern ist ausschließlich zum Inhalieren mit geeigneten Inhalationsgeräten bestimmt und darf nicht eingenommen oder parenteral angewendet werden.
Die Anwendung sollte möglichst im Sitzen oder Stehen erfolgen. Erläuterung zur Handhabung siehe Abschnitt 6.6.
4.3 Gegenanzeigen
Atrovent 500 pg/2 ml Fertiginhalat ist kontraindiziert bei Überempfindlichkeit gegen den Wirkstoff, Atropin oder anderen Atropinderivaten (wie z. B. Ipratropiumbromid), oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Überempfindlichkeit
Überempfindlichkeitsreaktionen vom Soforttyp können nach Anwendung von Atrovent 500 pg/2 ml Fertiginhalat aufreten, wie z. B. seltene Fälle von Exanthem, Urtikaria, Angioödem, oropharyngealem Ödem, Bronchospasmus und Anaphylaxie.
Paradoxer Bronchospasmus
Wie bei anderen Medikamenten zur Inhalation kann Atrovent 500 pg/2 ml Fertiginhalat paradoxe Bronchospasmen hervorrufen, die lebensbedrohlich sein können. Falls paradoxe Bronchospasmen aufreten, muss Atrovent 500 pg/2 ml Fertiginhalat unverzüglich abgesetzt und durch eine alternative Therapie ersetzt werden.
Besondere Patientengruppen:
Okulare Komplikationen
Es muss sorgfältig darauf geachtet werden, dass die Lösung oder der Inhalationsnebel nicht in die Augen gelangt.
Atrovent 500 pg/2 ml Fertiginhalat sollte bei Patienten mit einer Prädisposition für ein Engwinkelglaukom mit Vorsicht angewendet werden.
Wenn das Arzneimittel bei der Anwendung versehentlich in die Augen gelangt, können leichte und reversible Augenkomplikationen auftreten. Insbesondere bei Patienten mit Engwinkelglaukom besteht die Möglichkeit eines akuten Glaukomanfalls mit folgenden typischen Symptomen: Augenschmerzen, unscharfes Sehen, Augenhalos oder unwirkliches Farbempfinden, gerötete Augen und Corneaödeme. Sollten Pupillenerweiterung und leichte Akkommodationsstörungen eintreten, können diese mit miotischen Augentropfen behandelt werden. Beim Auftreten von schweren Augenkomplikationen sollte zusätzlich ein Augenarzt aufgesucht werden.
Bei diesen Patienten sollte möglichst ein Mundstück und keine Gesichtsmaske bei der Inhalation verwendet werden, damit das Arzneimittel nicht in die Augen gerät.
Wirkung auf Nieren und Harnwege
Bei Patienten mit Miktionsstörungen (z. B. bei Prostatahyperplasie oder Blasenhalsobstruktion) ist der Nutzen einer Ipratropiumbromid-Behandlung sorgfältig gegen das mögliche Risiko einer Verstärkung des Harnverhalts abzuwägen.
Störungen der gastrointestinalen Motilität
Bei Patienten mit zystischer Fibrose kann es eher zu gastrointestinalen Motilitätsstörungen kommen.
4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Die chronische Anwendung von Atrovent zur Inhalation zusammen mit anderen anticholinergen Arzneimitteln wurde nicht untersucht und wird deshalb nicht empfohlen.
ß-Adrenergika und Xanthinderivate (z. B. Theophyllin) können die Wirkung verstärken.
Andere Anticholinergika, wie z. B. pirenzepinhaltige Präparate, können Wirkung und Nebenwirkungen verstärken.
Das Risiko eines akuten Glaukomanfalls bei Patienten mit Engwinkelglaukom kann erhöht sein, wenn Atrovent 500 pg/2 ml Fertiginhalat und ß-Mimetika zusammen angewendet werden.
4.6 Fertilität, Schwangerschaft und Stillzeit
Schwangerschaft und Stillzeit
Es liegen keine Erfahrungen beim Menschen mit der Anwendung in Schwangerschaft und Stillzeit vor.
Obwohl bisher keine fruchtschädigenden Wirkungen bekannt sind, sollte Atrovent 500 pg/2 ml Fertiginhalat in der Schwangerschaft, insbesondere während des ersten Trimenons und während der Stillzeit, nur dann angewendet werden, wenn dies vom behandelnden Arzt nach sorgfältiger NutzenRisiko-Abwägung als notwendig erachtet wird.
Die Risiken einer unzureichenden Behandlung sollten dabei angemessen berücksichtigt werden. Fertilität
Klinische Daten zur Fertilität liegen für Ipratropiumbromid nicht vor. Nichtklinische Studien mit Ipratropiumbromid zeigten keine unerwünschte Wirkung auf die Fertilität (siehe Abschnitt 5.3).
4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Es wurden keine Studien zu Auswirkungen auf die Verkehrstüchtigkeit und auf die Fähigkeit zum Bedienen von Maschinen durchgeführt. Die Patienten sollten jedoch darauf hingewiesen werden, dass während der Behandlung mit Atrovent 500 pg/2 ml Fertiginhalat unerwünschte Wirkungen wie Schwindel, Akkommodationsstörungen, Mydriasis und unscharfes Sehen auftreten können. Deshalb sollte mit Vorsicht Auto gefahren oder Maschinen bedient werden.
4.8 Nebenwirkungen
Wie alle Arzneimittel kann Atrovent 500 pg/2 ml Fertiginhalat Nebenwirkungen haben.
a) Allgemeine Beschreibung
Viele der aufgeführten Nebenwirkungen können auf die anticholinergen Eigenschaften von Atrovent zurückgeführt werden.
b) Tabelle der Nebenwirkungen
Die aufgelisteten Nebenwirkungen basieren auf Daten aus klinischen Prüfungen und der Arzneimittelüberwachung der Anwendung nach der Zulassung. Bei der Bewertung von Nebenwirkungen werden folgende Häufigkeiten zugrunde gelegt:
|
Sehr häufig |
(> 1/10) |
|
Häufig |
(> 1/100, < 1/10) |
|
Gelegentlich |
(> 1/1.000, < 1/100) |
|
Selten |
(> 1/10.000, < 1/1.000) |
|
Sehr selten |
(< 1/10.000) |
|
Nicht bekannt |
(Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar) |
Erkrankungen des Immunsystems
Gelegentlich: anaphylaktische Reaktionen, Überempfindlichkeit
Erkrankungen des Nervensystems
Häufig: Kopfschmerzen, Schwindel
Augenerkrankungen
Gelegentlich: Verschwommenes Sehen, Mydriasis, Anstieg des
Augeninnendrucks ggf. mit Augenschmerzen, Sehen von Nebel und Regenbogenfarben (-ringen), Bindehauthyperämie und Hornhautödem, Glaukom Selten: Akkommodationsstörungen
Herzerkrankungen
Gelegentlich:
Selten:
Palpitationen, (supraventrikuläre) Tachykardien atriale Fibrillationen
Erkrankungen der Atemwege, des Brustraums und Mediastinums
Häufig: Husten, Rachen-Irritationen
Gelegentlich: (paradoxer) Bronchospasmus, Laryngospasmus,
Rachenödem, trockener Rachen
Erkrankungen des Gastrointestinaltrakts
Häufig: Trockener Mund, Geschmacksstörung,
gastrointestinale Motilitätsstörungen, Übelkeit
Gelegentlich: Verstopfung, Durchfall, Bauchschmerzen,
Erbrechen, Stomatitis, Mund-Ödem
Erkrankungen der Haut und des Unterhautzellgewebes
Gelegentlich: Hautausschlag, Pruritus, Angioödem
Selten: Urtikaria
Erkrankungen der Nieren und Harnwege
Gelegentlich: Harnverhalt
c) Angaben zu häufig auftretenden Nebenwirkungen
Wie bei jeder inhalativen Therapie können auch unter Atrovent Anzeichen von lokaler Reizung im Rachenbereich auftreten. In klinischen Prüfungen waren die am häufigsten berichteten Nebenwirkungen Kopfschmerz, Reizung des Rachens, Husten, trockener Mund, gastrointestinale Motilitätsstörungen (z. B. Verstopfung, Durchfall und Erbrechen), Übelkeit und Schwindel.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Arzneimittel und Medizinprodukte, Abt. Pharmakovigilanz, Kurt-Georg-Kiesinger-Allee 3, D-53175 Bonn, Website: www.bfarm.de anzuzeigen.
4.9 Überdosierung
Spezifische Überdosierungserscheinungen sind bisher nicht bekannt.
Durch die große therapeutische Breite und die lokale Applikation von Atrovent 500 qg/2 ml sind jedoch keine schweren anticholinergen Symptome zu erwarten. Leichte systemische anticholinerge Nebenwirkungen wie Mundtrockenheit, Akkommodationsstörungen der Augen und Anstieg der Herzfrequenz können auftreten.
5. PHARMAKOLOGISCHE EIGENSCHAFTEN
5.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Mittel bei obstruktiven Atemwegserkrankungen / Antiasthmatika /
Anticholinergika
ATC-Code: R03BB01
Ipratropiumbromid ist eine quaternäre Ammoniumverbindung mit anticholinergen (parasympatholytischen) Eigenschaften. Nichtklinische Studien zeigen eine Hemmung der vagal vermittelten Reflexe durch Antagonisierung der Wirkung von Acetylcholin, dem vom Nervus Vagus freigesetzten Transmitter. Anticholinergika verhindern die Zunahme der intrazellulären Ca++-Konzentration, die durch Interaktion von Acetylcholin mit dem Muscarinrezeptor auf der glatten Bronchialmuskelzelle verursacht wird. Die Freisetzung von Ca++ wird durch das Second Messenger System vermittelt, das aus IP3 (Inositoltriphosphat) und DAG (Diacylglycerol) besteht.
Die Bronchodilatation nach Inhalation mit Atrovent (Ipratropium) ist primär lokal und spezifisch an der Lunge und nicht systemischer Natur.
Präklinische und klinische Ergebnisse zeigen keinen negativen Effekt von Atrovent (Ipratropium) auf die Mucussekretion der Atemwege, die mukoziliäre Clearance oder den Gasaustausch.
Klinische Studien
In kontrollierten 85 - 90 Tages-Studien zeigten sich bei Patienten mit Bronchospasmen in Verbindung mit chronisch obstruktiver Lungenerkrankung (chronische Bronchitis und Emphysem) signifikante Verbesserungen der Lungenfunktion innerhalb von 15 Minuten, die einen Peak nach 1-2 Stunden erreichten und bis zu 4 bis 6 Stunden bestehen blieben.
Der bronchodilatorische Effekt von Atrovent in der Behandlung von akuten Bronchospasmen in Verbindung mit Asthma wurde in Studien mit Erwachsenen nachgewiesen. In den meisten dieser Studien wurde Atrovent zusammen mit einem inhalativen ß-Agonisten verabreicht.
5.2 Pharmakokinetische Eigenschaften
Resorption
Die therapeutische Wirksamkeit von Atrovent wird durch lokale Wirkung auf die Atemwege hervorgerufen. Die zeitliche Abfolge der Bronchodilatation und systemischen Pharmakokinetik verläuft nicht parallel.
Nach der Inhalation erfolgt in der Regel eine Deposition von 10 - 30 % der Dosis in der Lunge, je nach Formulierung und Inhalationstechnik. Der Großteil der Dosis wird geschluckt und über den Magen-Darm-Trakt ausgeschieden.
Der über die Lunge aufgenommene Dosisanteil geht schnell in den Blutkreislauf über (innerhalb von Minuten).
Die kumulative renale Exkretion (0 - 24 Std.) der Ausgangssubstanz liegt bei ca. 46 % bei einer intravenös verabreichen Dosis, unter 1 % bei einer oralen Dosis und bei ca. 3 - 13 % bei einer inhalierten Dosis. Auf Grundlage dieser Daten wird die gesamte systemische Bioverfügbarkeit oraler und inhalierter Dosen Ipratropiumbromid auf 2 % bzw. 7 - 28 % geschätzt.
Unter Berücksichtigung dieser Daten tragen geschluckte Dosisanteile von Ipratropiumbromid nicht erheblich zur systemischen Exposition bei.
Verteilung
Die kinetischen Parameter der Disposition von Ipratropium wurden aus Plasmakonzentrationen nach intravenöser Verabreichung errechnet. Eine schnelle biphasische Abnahme der Plasmakonzentrationen wurde beobachtet. Das scheinbare Verteilungsvolumen im Steady-State (Vdss) beträgt ca. 176 l (~ 2,4 l/kg). Der Wirkstoff bindet minimal (unter 20 %) an Plasmaproteine. Nichtklinische Daten deuten darauf hin, dass das quartäre Amin Ipratropium die Plazenta- und die Blut-Hirn-Schranke nicht überwindet.
Biotransformation
Nach intravenöser Verabreichung werden ca. 60 % einer Dosis metabolisiert, der größte Teil wahrscheinlich durch Oxidation in der Leber.
Die bekannten Metabolite, die durch Hydrolyse, Dehydratisierung oder Eliminierung der Hydroxylmethyl-Gruppe im Tropasäure-Anteil entstehen, binden schlecht an den Muscarinrezeptor und müssen als ineffektiv betrachtet werden.
Elimination
Die terminale Eliminationshalbwertszeit beträgt ca. 1,6 Stunden.
Ipratropium weist eine Gesamt-Clearance von 2,3 l/min und eine renale Clearance von 0,9 l/min auf.
Nach Inhalation von Ipratropiumbromid mit HFA 134a als Treibmittel betrug die kumulative renale Exkretion über 24 Stunden ca. 12 %.
In einer Studie zur Ausscheidungsbilanz betrug die kumulative renale Exkretion (6 Tage) der wirkstoffbezogenen Radioaktivität (einschließlich Ausgangssubstanz und sämtliche Metaboliten) nach intravenöser Gabe 72,1 %, nach oraler Verabreichung 9,3 % und nach Inhalation 3,2 %. Die Gesamtradioaktivität, die über die Faeces ausgeschieden wurde, betrug 6,3 % nach intravenöser Verabreichung, 88,5 % nach oraler Gabe und 69,4 % nach Inhalation. Die Exkretion der wirkstoffbezogenen Radioaktivität nach intravenöser Verabreichung erfolgt hauptsächlich über die Nieren. Die Eliminationshalbwertszeit der wirkstoffbezogenen Radioaktivität (Ausgangssubstanz und Metaboliten) beträgt 3,6 Stunden.
5.3 Präklinische Daten zur Sicherheit
Die lokale und systemische Verträglichkeit von Ipratropiumbromid wurden an mehreren Tierarten und über verschiedene Verabreichungswege umfassend geprüft.
Toxizität, nach Einmalgabe
Die akute inhalative, orale und intravenöse Toxizität wurde in mehreren Nagetier- und NichtNagetierarten untersucht.
Bei der inhalativen Verabreichung betrug die niedrigste letale Dosis bei männlichen Meerschweinchen 199 mg/kg. Bei Ratten wurde bis zu den höchsten technisch durchführbaren Dosierungen (d. h.
0,05 mg/kg nach 4-stündiger Verabreichung oder 160 Sprühstößen Ipratropiumbromid,
0,02 mg/Sprühstoß) keine Mortalität verzeichnet.
Der orale LD50-Wert betrug bei Mäusen 1585 mg/kg, bei Ratten 1925 mg/kg und bei Kaninchen 1920 mg/kg. Der intravenöse LD50-Wert betrug bei Mäusen 13,6 mg/kg, bei Ratten 15,8 mg/kg und bei Hunden ca. 18,2 mg/kg. Die klinischen Symptome umfassten Mydriasis, trockene Mundschleimhaut, Dyspnoe, Tremor, Krämpfe und/oder Tachykardie.
Toxizität nach Mehrfachgabe
Toxizitätsstudien mit wiederholter Wirkstoffgabe wurden mit Ratten, Kaninchen, Hunden und Rhesusaffen durchgeführt.
In Inhalationsstudien mit einer Dauer von bis zu sechs Monaten betrug der NOAEL (No Observed Adverse Effect Level) bei Ratten 0,38 mg/kg/Tag, bei Hunden 0,18 mg/kg/Tag und bei Rhesusaffen 0,8 mg/kg/Tag. Bei Hunden traten trockene Mundschleimhaut und Tachykardie auf. Im bronchopulmonalen System oder anderen Organen wurden keine wirkstoffbezogenen histopathologischen Läsionen beobachtet. Bei Ratten betrug der NOAEL nach 18 Monaten der oralen Verabreichung 0,5 mg/kg/Tag.
Studien zur Inhalationstoxizität mit wiederholter Wirkstoffgabe, bei der andere Formulierungen (intranasale Formulierung, alternatives Treibmittel HFA 134a und Laktosepulverformulierungen) bis zu sechs Monate an Ratten und bis zu drei Monate an Hunden getestet wurden, ergaben keine weiteren Informationen zum allgemeinen Toxizitätsprofil von Ipratropiumbromid.
Die intranasale Verabreichung über bis zu sechs Monate ergab einen NOEL (No Effect Level) von > 0,20 mg/kg/Tag bei Hunden und bestätigte frühere Studien zur intranasalen Verabreichung über bis zu 13 Wochen. Toxizitätsstudien mit wiederholter Wirkstoffgabe von Ipratropiumbromid haben gezeigt, dass die toxikologischen Profile der HFA-Formulierung und der konventionellen CFC-Formulierung ähnlich sind.
Lokale Verträglichkeit
Eine wässrige Lösung von Ipratropiumbromid (0,05 mg/kg) wurde bei inhalativer Verabreichung von Ratten lokal gut vertragen (eine Verabreichung in vier Stunden). In der Toxizitätsstudie mit wiederholter Wirkstoffgabe wurde Ipratropiumbromid lokal gut vertragen.
Immunogene Aktivität
Bei Meerschweinchen traten weder eine aktive Anaphylaxie noch passive anaphylaktische Reaktionen der Haut auf.
Genotoxizität und Karzinogenizität
In vitro (Ames-Test) und in vivo (Mikronukleustest, Dominant-Letal-Test bei Mäusen, zytogenetischer Assay mit Rückenmarkszellen chinesischer Hamster) wurden keine Nachweise für genotoxische Effekte erbracht.
In Langzeitstudien mit Mäusen und Ratten wurden keine tumorigenen oder kanzerogenen Effekte festgestellt.
Reproduktions- und Entwicklungstoxizität
Studien zur Untersuchung des potenziellen Einflusses von Ipratropiumbromid auf Fertilität, Embryo-/Fetotoxizität sowie auf die peri-/postnatale Entwicklung wurden bei Mäusen, Ratten und Kaninchen durchgeführt.
Hohe orale Dosen, d. h. 1000 mg/kg/Tag bei Ratten und 125 mg/kg/Tag bei Kaninchen, wirkten bei beiden Tierarten maternotoxisch und bei Ratten embryo-/fetotoxisch und führten zu geringerem Fetalgewicht. Fehlbildungen in Zusammenhang mit der Wirkstoffgabe wurden nicht beobachtet.
Die höchsten technisch durchführbaren Inhalationsdosen des Dosieraerosols - 1,5 mg/kg/Tag bei Ratten und 1,8 mg/kg/Tag bei Kaninchen - hatten keine unerwünschten Wirkungen auf die Reproduktion.
6. PHARMAZEUTISCHE ANGABEN
6.1 Liste der sonstigen Bestandteile
Natriumchlorid, Salzsäure 3,6 %-ig (zur pH-Einstellung), gereinigtes Wasser
6.2 Inkompatibilitäten
Atrovent 500 ^g/2 ml Fertiginhalat und DNCG (Dinatriumcromoglycat)-Lösungen sollen nicht gleichzeitig im selben Vernebler inhaliert werden.
6.3 Dauer der Haltbarkeit
2 Jahre
6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Nicht über 30 °C lagern, in der Originalschachtel aufbewahren.
6.5 Art und Inhalt des Behältnisses
Klare, farblose Lösung für einen Vernebler in einem farblosen, transparenten Eindosisbehälter aus Kunststoff (PE). 10 Eindosisbehälter sind jeweils in einem hermetisch abgeschlossenen weißen Aluminiumbeutel verpackt.
Packungsgrößen:
Originalpackung mit 50 Eindosisbehälter a 2 ml
6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung
Diese Lösung ist gebrauchsfertig, d. h. eine Verdünnung ist nicht erforderlich.
Die Lösung in den Eindosisbehältern ist ausschließlich zum Inhalieren mit geeigneten Inhalationsgeräten bestimmt, darf nicht eingenommen oder parenteral angewendet werden.
Die Anwendung sollte möglichst im Sitzen oder Stehen erfolgen.
Gebrauchsanweisung
1. Inhalationsgerät (Vernebler) nach den Anweisungen des Herstellers und des behandelnden Arztes zur Inhalation vorbereiten.
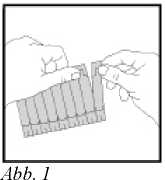
2. Einen Eindosisbehälter von dem Streifen abtrennen (s. Abb. 1).
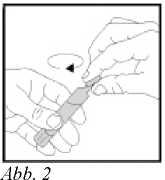
3. Zum Öffnen des Eindosisbehälters den oberen Teil (die Abrissfahne) abdrehen (s. Abb. 2).
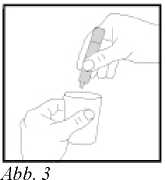
4. Inhalt durch mehrmaliges Zusammendrücken des Eindosisbehälters in den Vemeblerbehälter geben (s. Abb. 3).
5. Vernebler zusammensetzen und nach entsprechender Anweisung inhalieren.
6. Nach der Inhalation die eventuell verbleibende Lösung aus dem Verneblerbehälter entfernen und den Vernebler wie vorgeschrieben säubern.
Die Patienten müssen hinsichtlich der korrekten Anwendung von Atrovent 500 ^g/2 ml Fertiginhalat instruiert werden.
Es muss sorgfältig darauf geachtet werden, dass die Lösung oder der Inhalationsnebel nicht in die Augen gelangt. Die vernebelte Lösung sollte durch ein Mundstück inhaliert werden. Wenn kein Mundstück verfügbar ist und eine Verneblungsmaske verwendet wird, muss auf deren korrekten Sitz geachtet werden. Patienten mit Neigung zu Glaukom sollen besonders darauf achten, dass ihre Augen während der Inhalation geschützt sind.
Da die Lösung keine Konservierungsmittel enthält, muss der Inhalt des Eindosisbehälters unmittelbar nach dem Öffnen inhaliert werden, eventuell verbleibende Restlösung ist zu verwerfen.
Für jede Anwendung ist ein neuer Eindosisbehälter zu verwenden. Angebrochene, versehentlich geöffnete oder beschädigte Eindosisbehälter sind zu verwerfen.
Atrovent 500 ^g/2 ml Fertiginhalat kann mit den handelsüblichen Verneblergeräten verwendet werden; bei Sauerstoffzufuhr ist die beste Verabreichung mit einem Fluss von 6 - 8 l/min.
Atrovent 500 ^g/2 ml Fertiginhalat ist zur gleichzeitigen Inhalation mit Ambroxol, z. B. enthalten in Mucosolvan® Inhalationslösung, geeignet.
7. INHABER DER ZULASSUNG
Boehringer Ingelheim Pharma GmbH & Co. KG
Binger Str. 173
55216 Ingelheim am Rhein
Telefon: 0 800 / 77 90 900
Telefax: 0 61 32 / 72 99 99
E-Mail: info@boehringer-ingelheim.de
8. ZULASSUNGSNUMMER(N)
37146.00.00
9. DATUM DER ERTEILUNG/VERLÄNGERUNG DER ZULASSUNG
Datum der Erteilung der Zulassung: 09.07.1998 Datum der Verlängerung der Zulassung: 10.02.2004
10. STAND DER INFORMATION
August 2016
11. VERKAUFSABGRENZUNG
Verschreibungspflichtig
Seite 10 von 10