Durafenat Retard
Fachinformation
1. Bezeichnung des Arzneimittels
durafenat retard Hartkapseln, retardiert
2. Qualitative und quantitative Zusammensetzung
Wirkstoff: Fenofibrat
1 Hartkapsel, retardiert durafenat retard enthält 250 mg Fenofibrat.
Sonstige Bestandteile:
Sucrose
Vollständige Auflistung der sonstigen Bestandteile siehe Abschnitt 6.1.
3. Darreichungsform
Hartkapsel, retardiert
4. Klinische Angaben
4.1 Anwendungsgebiete
durafenat retard ist angezeigt als unterstützende Behandlung neben einer Diät oder anderen nicht-medikamentösen Therapien (z. B. sportlicher Betätigung, Gewichtsabnahme) für folgende Erkran-kungen:
- schwere Hypertriglyceridämie mit oder ohne niedrige HDL-Cholesterinwerte
- gemischte Hyperlipidämie, wenn ein Statin kontraindiziert ist oder nicht vertragen wird
- bei gemischter Hyperlipidämie bei Patienten mit hohem kardiovaskulären Risiko zusätzlich zu einem Statin, wenn Triglyzerid- und HDL-Cholesterinwerte nicht ausreichend kontrolliert werden können.
4.2 Dosierung und Art der Anwendung
Am Anfang jeder Behandlung einer Hyperlipidämie stehen immer eine Ernährungsberatung und die Identifizierung und Korrektur von Risikofaktoren.
In vielen Fällen sind Fettstoffwechselstörungen durch diätetische Maßnahmen, Gewichtsreduktion, vermehrte körperliche Aktivität und durch ausreichende Behandlung gleichzeitig bestehender anderer Stoffwechselerkrankungen günstig zu beeinflussen. Diese Maßnahmen sollten während der Behandlung mit durafenat retard beibehalten werden.
Bei der Diagnosestellung ist zu berücksichtigen, dass der Blutlipidspiegel von verschiedenen Faktoren wie Tageszeit, Abstand vom Zeitpunkt der Einnahme und Beschaffenheit der letzten Mahlzeit, Alkoholgenuss, Stresssituation abhängig ist. Bei hyperlipämischen Patienten, die Östrogene oder östrogenhaltige Kontrazeptiva einnehmen, sollte geprüft werden, ob es sich um eine primäre oder sekundäre Hyperlipidämie handelt (möglicher Anstieg der Lipidwerte durch Östrogene).
Da die medikamentöse Therapie der Hyperlipidämie meistens eine Langzeitbehandlung bedeutet, muss die Entscheidung zur Einleitung einer solchen Therapie im Einzelfall sorgfältig abgewogen werden.
Dosierung:
1 Hartkapsel, retardiert (entsprechend 250 mg Fenofibrat) täglich.
Ältere Patienten (ab 65 Jahren):
Eine Dosisanpassung ist nicht notwendig. Die Einnahme der üblichen Dosis wird empfohlen, außer bei eingeschränkter Nierenfunktion mit einer geschätzten (estimated) glomerulären Filtrationsrate (eGFR) < 60 ml/min/1,73 m2 (siehe „Patienten mit eingeschränkter Nierenfunktion“).
Patienten mit eingeschränkter Nierenfunktion:
Bei stark eingeschränkter Nierenfunktion mit einer eGFR < 30 ml/min/1,73 m2 darf Fenofibrat nicht eingenommen werden.
Bei einer eGFR zwischen 30 und 59 ml/min/1,73 m2 sollte die tägliche Dosis 100 mg Fenofibrat (Standard) oder 67 mg mikronisiert nicht überschreiten. Wenn in Nachuntersuchungen die eGFR dauerhaft unter 30 ml/min/1,73 m2 fällt, muss die Einnahme von Fenofibrat abgebrochen werden.
Kinder und Jugendliche
Die Unbedenklichkeit und Wirksamkeit von Fenofibrat bei Kindern und Jugendlichen unter 18 Jahren ist nicht hinreichend nachgewiesen. Es liegen keine Studien vor. Aus diesem Grund wird die Anwendung von Fenofibrat bei Kindern und Jugendlichen unter 18 Jahren nicht empfohlen.
Art der Anwendung:
Die Hartkapseln, retardiert sollen unzerkaut mit etwas Flüssigkeit zum Essen eingenommen werden.
4.3 Gegenanzeigen
- Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile, photoallergische oder phototoxische Reaktionen unter einer Behandlung mit Fibraten oder Ketoprofen,
- Lebererkrankungen (mit Ausnahme der Fettleber, die häufiges Begleitsymptom bei
Hypertriglyzeridämie ist)
- schwere Niereninsuffizienz (geschätzte glomeruläre Filtrationsrate < 30 ml/min/1,73 m2)
Als relative Kontraindikation gelten Gallenblasenerkrankungen mit und ohne Cholelithiasis, da die Möglichkeit einer Leberbeteiligung nicht ausgeschlossen werden kann.
durafenat retard ist für Kinder nicht geeignet.
Weitere Informationen siehe 4.4. Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung.
4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Nierenfunktion:
durafenat retard ist kontraindiziert bei stark eingeschränkter Nierenfunktion (siehe Abschnitt 4.3).
durafenat retard sollte bei Patienten mit leichter Niereninsuffizienz mit Vorsicht angewendet werden. Bei Patienten mit einer eGFR zwischen 30 und 59 ml/min/1,73 m2 sollte durafenat retard aufgrund des hohen Wirkstoffgehaltes nicht angewendet werden (siehe Abschnitt 4.2).
Reversible Kreatininwerterhöhungen im Blut wurden bei Patienten, die eine Fenofibrat-Monotherapie oder eine Kombination mit Statinen erhalten haben, beobachtet. Die Kreatininwerterhöhung war im Allgemeinen über die Zeit stabil, Anzeichen eines weiteren Anstiegs wurden bei einer Langzeittherapie nicht beobachtet. Nach Beendigung der Behandlung wurde ein Rückgang auf die Ausgangswerte beobachtet.
In klinischen Studien hatten 10 % der Patienten bei der Kombinationsbehandlung von Fenofibrat und Simvastatin einen auf die Ausgangswerte bezogenen Kreatininanstieg um mehr als 30 ^mol/l im Vergleich zu 4,4 % der Patienten bei der Statin-Monotherapie. 0,3 % der Patienten, die die Kombinationsbehandlung erhielten, hatten klinisch relevante Anstiege von Kreatinin auf Werte größer 200 ^mol/l.
Die Behandlung sollte abgebrochen werden, wenn der Kreatininwert den oberen Normwert um 50% übersteigt. Es wird empfohlen, den Kreatininwert während der ersten drei Monate nach Therapiebeginn und danach in periodischen Abständen zu kontrollieren.
Bei der Anwendung von Fibraten und anderen Lipidsenkern wurde über Myotoxizität und in sehr seltenen Fällen über Rhabdomyolyse berichtet. Bei Patienten mit Hypalbuminämie und Niereninsuffizienz in der Vorgeschichte ist die Inzidenz von Myotoxizität erhöht. Diffuse Myalgien, Myositis, Muskelkrämpfe, Muskelschwäche und/ oder ein erheblicher Anstieg der Kreatin-Phosphokinase (CPK) (Anstieg über das Fünffache des Normwertes) deuten auf eine Myotoxizität hin. Das Arzneimittel ist in diesen Fällen abzusetzen.
Das Risiko einer Myotoxizität kann sich erhöhen, wenn dieses Arzneimittel zusammen mit einem anderen Fibrat oder einem HMG-CoA-Reduktasehemmer kombiniert wird. Dies gilt insbesondere, wenn bereits Muskelerkrankungen bestehen. Daher sollte die Kombination von Fenofibrat mit einem Statin auf Patienten beschränkt werden mit schwerer kombinierter Hyperlipidämie und hohem kardiovaskulärem Risiko, bei denen bislang noch keine Muskelerkrankungen aufgetreten sind. Diese Kombinationstherapie sollte mit Vorsicht eingesetzt werden und die Patienten sollten streng auf eine mögliche Myotoxizität hin überwacht werden.
Dieses Arzneimittel enthält Sucrose. Patienten mit der seltenen hereditären FructoseIntoleranz, Glucose-Galactose-Malabsorption oder Saccharase-Isomaltase-Mangel sollten durafenat retard nicht einnehmen.
4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Antikoagulantien:
Fenofibrat kann die Wirkung der Antikoagulantien vom Cumarin-Typ verstärken. Deshalb sollte zu Beginn einer Therapie mit durafenat retard die Phenprocoumon-Dosis um ca. 30 % reduziert und unter Kontrolle der Blutgerinnung neu eingestellt werden. Auch nach Absetzen von durafenat retard ist eine Neueinstellung erforderlich.
Antidiabetika:
Die Wirkung von oralen blutzuckersenkenden Arzneimitteln und Insulin kann durch durafenat retard verstärkt werden.
Fenofibrat soll wie andere Fibrate wegen der Gefahr einer Rhabdomyolyse nicht mit HMG-CoA-Reduktasehemmern (Cholesterolsynthesehemmern) oder anderen Fibraten kombiniert werden (siehe 8. Warnhinweise und Vorsichtsmaßnahmen für die Anwendung).
In Einzelfällen wurde bei organtransplantierten Patienten unter immunsuppressiver Therapie bei gleichzeitiger Anwendung von Fibrat-haltigen Arzneimitteln über eine erhebliche, wenn auch reversible Einschränkung der Nierenfunktion (mit entsprechendem Anstieg des Serumkreatinins) berichtet. Daher ist bei diesen Patienten die Nierenfunktion sorgfältig zu überwachen, und bei diesbezüglich bedeutsamen Veränderungen der labordiagnostischen Parameter ist durafenat retard ggf. abzusetzen.
4.6 Fertilität, Schwangerschaft und Stillzeit
durafenat retard darf in der Schwangerschaft und Stillzeit nicht angewendet werden, da keine Erfahrungen mit der Anwendung in Schwangerschaft und Stillzeit beim Menschen vorliegen (s. a. Punkt 5.3 Präklinische Daten zur Sicherheit).
4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Nicht zutreffend.
4.8 Nebenwirkungen
Bei der Bewertung von Nebenwirkungen werden folgende Häufigkeiten zugrunde gelegt:
Sehr häufig (>1/10)
Häufig (>1/100 bis <1/10)
Gelegentlich (>1/1.000 bis <1/100)
Selten (>1/10.000 bis <1/1.000)
Sehr selten (<1/10.000)
Nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar)
Folgende Nebenwirkungen können unter der Behandlung mit durafenat retard auftreten:
Erkrankungen des Blutes und des Lymphsystems:
Über leichte Abnahme von Hämoglobin und Leukozyten wurde berichtet.
Erkrankungen des Nervensystems:
In einzelnen Fällen ist über Potenzstörungen berichtet worden. Diese Nebenwirkung klingt im Allgemeinen nach Absetzen von durafenat retard rasch ab.
Erkrankungen des Gastrointestinaltrakts:
Gastrointestinale Störungen (wie Völlegefühl, Übelkeit, Obstipation; Diarrhoe) sind im Allgemeinen vorübergehend und erfordern kein Absetzen der Medikation.
Leber-Gallenerkrankungen:
Während der Behandlung mit durafenat retard kann es zu einem vorübergehenden Anstieg bestimmter Leberenzyme (SGOT und SGPT) kommen.
In Einzelfällen wurde unter Fenofibrat eine cholestatische Hepatitis beobachtet, die nach Absetzen von Fenofibrat reversibel war. Bei Auftreten von Symptomen (z. B. Ikterus, Juckreiz), die auf eine cholestatische Hepatitis hinweisen, ist die Kontrolle der erforderlichen labordiagnostischen Parameter durchzuführen und durafenat retard ggf. abzusetzen. I
Infolge der vermehrten Cholesterinausscheidung unter der Behandlung mit durafenat retard erhöht sich der lithogene Index (der ein Maß für die Cholesterinsättigung der Galle ist) und damit das mögliche Gallensteinrisiko. Ob unter Langzeitbehandlung vermehrt Gallensteine auftreten oder vorhandene Gallensteine an Größe zunehmen, ist umstritten.
Erkrankungen der Haut und des Unterhautzellgewebes:
Gelegentlich können allergische Reaktionen wie Pruritus, Urtikaria oder andere Hauterscheinungen auftreten, die nach Absetzen von durafenat retard verschwinden.
In Einzelfällen kann es - auch nach monatelanger komplikationsloser Anwendung - zu im Allgemeinen reversiblen photoallergischen oder phototoxischen Reaktionen mit Erythem, Pruritus, Bläschenbildung oder lichenoiden Veränderungen kommen. durafenat retard ist in diesem Fall sofort abzusetzen.
In Einzelfällen ist über Haarausfall berichtet worden. Diese Nebenwirkung klingt im Allgemeinen nach Absetzen von durafenat retard rasch ab.
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen:
Eine wichtige, jedoch seltene Nebenwirkung ist eine Myotoxizität mit Muskelschmerzen, Muskelschwäche und Muskelkrämpfen; in diesem Fall sollte eine Bestimmung der Kreatin-Phosphokinase (CPK) erfolgen. Sehr selten kann ein erheblicher CPK-Anstieg mit dem klinischen Bild einer medikamentös bedingten Rhabdomyolyse auftreten; dem liegt häufig eine zu hohe Dosierung z. B. durch Kumulation bei Niereninsuffizienz, zugrunde (siehe Dosierungsanleitung). Das Arzneimittel ist in diesen Fällen sofort abzusetzen (siehe 4.4. Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung)
Untersuchungen:
Unter Langzeitbehandlung kommt es zu einem leichten Anstieg des Serumkreatinins und des Serumharnstoffes.
4.9 Überdosierung
Ein spezielles Antidot besteht nicht. Bei Verdacht auf Überdosierung und Rhabdomyolyse ist die Medikation abzubrechen. Bei Nierengesunden kann durch forcierte Diurese versucht werden, die Elimination zu beschleunigen. Bei Rhabdomyolyse ist durch ausreichende Flüssigkeitsgabe der Entstehung einer Crushniere vorzubeugen. Fenofibrat ist nicht dialysierbar.
5. PHARMAKOLOGISCHE EIGENSCHAFTEN
5.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Lipidsenker ATC-Code: C10AB05
Fenofibrat senkt erhöhte Blutlipide (Triglyzeride und Cholesterin) und dient deshalb zur Therapie bestimmter Hyperlipoproteinämien. Der Mechanismus der Lipidsenkung beruht überwiegend auf einer Aktivierung lipolytischer Enzyme, insbesondere der Lipoproteinlipase (LPL), wodurch es zu einem beschleunigten Katabolismus triglyceridreicher very-low-density-Lipoproteine (VLDL) und zu einem parallelen Anstieg antiatherogener high-density-Lipoproteine (HDL) kommt.
Es liegen Belege dafür vor, dass die Behandlung mit Fibraten die Häufigkeit von Ereignissen bei koronaren Herzerkrankungen reduziert. Es liegen jedoch keine Hinweise für einen positiven Effekt im Hinblick auf die Gesamtmortalität in der primären oder sekundären Vorbeugung kardiovaskulärer Erkrankungen vor.
Bei der ACCORD (Action to Control Cardiovascular Risk in Diabetes) Lipid-Studie handelte es sich um eine randomisierte placebokontrollierte Studie bei 5.518 Patienten mit Typ-2-Diabetes mellitus, die zusätzlich zu Simvastatin mit Fenofibrat behandelt wurden. Bei der Behandlung mit Fenofibrat plus Simvastatin wurden gegenüber der Simvastatin-Monothe-rapie keine signifikanten Unter-schiede hinsichtlich des kombinierten primären Endpunkts bestehend aus nicht-tödlichem Myo-kardinfarkt, nicht-tödlichem Schlaganfall und kardiovaskulär bedingtem Tod beobachtet (Hazard Ratio [HR] 0,92; 95 %-Konfidenzintervall:
0,79 bis 1,08; p = 0,32; absolute Risikoreduktion: 0,74 %). In der vorab festgelegten Untergruppe dyslipidämischer Patienten, definiert als diejenigen Patienten in der untersten Tertile des HDL-C-Werts (< 34 mg/dl bzw. 0,88 mmol/l) und in der obersten Tertile des TG-Werts (> 204 mg/dl bzw. 2,3 mmol/l), wurde bei der Behandlung mit Feno-fibrat plus Simvastatin gegenüber der Simvastatin-Monotherapie eine relative Risikoreduktion von 31 % in Bezug auf das kombinierte primäre Zielkriterium beobachtet (Hazard Ratio [HR] 0,69; 95 %-Kon-fidenzintervall: 0,49 bis 0,97; p = 0,03 ; absolute Risikoreduktion: 4,95 %). Eine weitere vorab festgelegte Untergruppenanalyse ergab eine statistisch signifikante geschlechtsspezifische Interaktion bei der Behandlung (p = 0,01), die auf einen möglichen Behandlungsnutzen der Kombinationstherapie bei Männern hinweist (p = 0,037), während bei Frauen für die Kombinationstherapie im Vergleich zur Simvastatin-Monotherapie ein potentiell höheres
Risiko für das Erreichen des primären Endpunkts bestand (p = 0,069). In der bereits genannten Untergruppe dyslipidämischer Patienten wurde eine solche Interaktion nicht beobachtet, es gab jedoch keine klaren Belege für den Nutzen einer Behandlung dyslipidämischer Frauen mit Fenofibrat plus Simvastatin; ferner konnte in dieser Untergruppe eine mögliche nachteilige Wirkung nicht ausgeschlossen werden.
5.2 Pharmakokinetische Eigenschaften
Nach oraler Applikation wird Fenofibrat schnell und nahezu vollständig resorbiert und zu Fenofibrinsäure hydrolysiert. Bei gesunden Probanden wird nach Einnahme einer Einzeldosis von 300 mg ein maximaler Plasmaspiegel von 15 mg/l erreicht. Fenofibrinsäure ist im Plasma zu 99 % an Proteine, vorwiegend Albumin, gebunden.
Die Ausscheidung von Fenofibrinsäure erfolgt zu ca. 60 % mit dem Urin und zu ca. 25 % mit den Fäces. Die Elimination ist zweiphasig, die Halbwertzeit der Alpha-Phase beträgt 5 Stunden, die der Beta-Phase 22 Stunden.
Bei Patienten mit Niereninsuffizienz ist die Ausscheidung reduziert, deshalb muss in diesem Fall die Dosis entsprechend der Serumkreatininclearance verringert werden (siehe 10. Dosierungsanleitung); bei Patienten mit leicht eingeschränkter Leberfunktion ist eine verzögerte Ausscheidung nicht bekannt (Lebererkrankungen siehe 5. Gegenanzeigen).
5.3 Präklinische Daten zur Sicherheit
a) Akute Toxizität
(siehe 4.9. Überdosierung)
b) Chronische Toxizität
Untersuchungen zur chronischen Toxizität ergaben keine relevanten Hinweise auf eine spezifische Toxizität von durafenat retard.
c) Mutagenes und tumorerzeugendes Potential
Untersuchungen zur Mutagenität von durafenat retard verliefen negativ.
Bei Ratten und Mäusen wurden in hohen Dosen Lebertumoren gefunden, die auf Peroxisomenproliferation zurückzuführen sind. Diese Veränderungen sind spezifisch für kleine Nager und wurden bei anderen Tierarten nicht beobachtet. Eine Relevanz für die therapeutische Anwendung beim Menschen ergibt sich daraus nicht.
d) Reproduktionstoxikologie
Untersuchungen an Maus, Ratte und Kaninchen ergaben keine Hinweise auf eine teratogene Wirkung. Embryotoxische Effekte wurden bei Dosierungen beobachtet, die im maternaltoxischen Bereich liegen. In hohen Dosen traten Tragzeitverlängerungen und eine Beeinträchtigung des Geburtsvorgangs auf. Hinweise auf eine Beeinflussung der Fertilität ergaben sich nicht. Es liegen keine Erfahrungen beim Menschen mit der Anwendung in der Schwangerschaft und in der Stillzeit vor.
6. PHARMAZEUTISCHE ANGABEN
6.1 Liste der sonstigen Bestandteile
Sucrose, Maisstärke, Talkum, Methacrylsäure-Methylmethacrylat-Copolymer (1:1) (Ph.Eur.) ((MW: ca. 135000)), basisches Butylmethacrylat-Copolymer (Ph.Eur.) ((MW: ca. 150000)), Gelatine, Titandioxid (E 171).
6.2 Inkompatibilitäten
Nicht zutreffend.
6.3 Dauer der Haltbarkeit
Die Dauer der Haltbarkeit beträgt 3 Jahre.
6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Für dieses Arzneimittel sind keine besonderen Lagerungsbedingungen erforderlich
6.5 Art und Inhalt des Behältnisses
OP mit 30 Hartkapseln, retardiert OP mit 50 Hartkapseln, retardiert OP mit 100 Hartkapseln, retardiert
6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung
Keine besonderen Anforderungen.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu entsorgen.
7. Inhaber der Zulassung
Mylan dura GmbH Postfach 10 06 35 64206 Darmstadt
8. Zulassungsnummer(n)
8307.00.00
Datum der Erteilung der Zulassung / Verlängerung der Zulassung
9.
30.06.1986 / 22.12.2004
10. Stand der Information
September 2016
11. Verkaufsabgrenzung
Verschreibungspflichtig
Bioverfügbarkeit
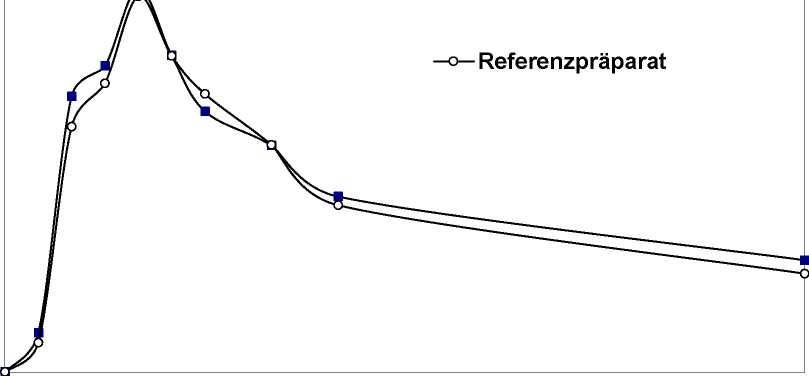
Eine im Jahr 1989 durchgeführte Bioverfügbarkeitsuntersuchung an 12 Probanden ergab für durafenat retard im Vergleich zum Referenzpräparat:
|
durafenat retard |
Referenzpräparat [1 x 250 mg] | |
|
maximale Plasmakonzentration |
6,64 ± 0,43 |
6,49 ± 0,43 |
|
Cmax in ^g / ml: | ||
|
Fläche unter der Konzentrations- |
74,91 ± 8,88 |
71,48 ± 10,80 |
|
Zeit-Kurve AUC (0-t) in ^g/ml x h: |
Angabe der Werte als Mittelwerte und Streubreite
Mittlere Plasmaspiegelverläufe im Vergleich zu einem Referenzpräparat in einem KonzentrationsZeit-Diagramm:
--durafenat retard
Plasmakonz. [ng/ml]
Zeit [h]