Fibclot
Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden. Hinweise zur Meldung von Nebenwirkungen, siehe Abschnitt 4.8.
1. BEZEICHNUNG DES ARZNEIMITTELS
FibCLOT 1,5 g. Pulver und Lösungsmittel zur Herstellung einer Injektions- bzw. Infusionslösung.
2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
Humanes Fibrinogen
Jede Durchstechflasche FibCLOT enthält nominal 1,5 g humanes Fibrinogen.
Nach Rekonstitution mit 100 ml Lösungsmittel (Wasser zu Injektionszwecken) enthält FibCLOT 15 mg/ml humanes Fibrinogen.
Die Aktivität wird entsprechend der Monografie des Europäischen Arzneibuchs für humanes Fibrinogen bestimmt.
Hergestellt aus dem Plasma menschlicher Spender.
Sonstige Bestandteile mit bekannter Wirkung:
Das Produkt enthält maximal 69 mg Natrium/Durchstechflasche.
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
3. DARREICHUNGSFORM
Pulver und Lösungsmittel zur Herstellung einer Injektions- bzw. Infusionslösung. Weißes oder hellgelbes Pulver in einer Durchstechflasche.
4 KLINISCHE ANGABEN
4.1. Anwendungsgebiete
Behandlung und perioperative Prophylaxe von Blutungen bei Patienten mit kongenitaler Hypo- oder Afibrinogenämie mit Blutungsneigung.
4.2. Dosierung und Art der Anwendung
Die Behandlung sollte unter der Überwachung eines Arztes erfolgen, der über Erfahrung in der Behandlung von Blutgerinnungsstörungen verfügt.
Dosierung
Dosierung und Dauer der Substitutionstherapie hängen von der Schwere der Erkrankung, der Lokalisation und dem Umfang der Blutung sowie vom Gesundheitszustand des Patienten ab.
Die individuelle Dosierung sollte auf Grundlage des (funktionalen) Fibrinogenspiegels berechnet werden. Menge und Häufigkeit der Anwendung sollten individuell für den einzelnen Patienten durch regelmäßige Messung des Fibrinogenspiegels im Plasma und durch kontinuierliche Überwachung des klinischen Zustands des Patienten und anderer verwendeter Substitutionstherapien bestimmt werden.
Der normale Fibrinogenspiegel im Plasma liegt zwischen 1,5-4,5 g/l. Bei kongenitaler Hypo- oder Afibrinogenämie liegt der kritische Fibrinogenspiegel im Plasma, unterhalb dessen es zu Blutungen kommen kann, bei circa 0,5-1,0 g/l.
Bei größeren chirurgischen Eingriffen ist eine präzise Überwachung der Substitutionstherapie durch Gerinnungstests unbedingt erforderlich.
Behandlung von Blutungen und Prophylaxe bei Patienten mit kongenitaler Hypo- oder Afibrinogenämie und bekannter Blutungsneigung.
Zur Behandlung nicht-chirurgischer Blutungsepisoden wird empfohlen, den Fibrinogenspiegel auf 1 g/l anzuheben und das Fibrinogen auf diesem Wert zu halten, bis die Hämostase unter Kontrolle ist, und auf über 0,5 g/l zu halten, bis eine vollständige Heilung erreicht ist.
Um übermäßige Blutungen während chirurgischer Eingriffe zu verhindern, wird eine prophylaktische Behandlung empfohlen. Der Fibrinogenspiegel ist auf 1 g/l anzuheben und auf diesem Wert zu halten, bis die Hämostase unter Kontrolle ist, und auf über 0,5 g/l zu halten, bis die Wundheilung abgeschlossen ist.
Für einen chirurgischen Eingriff oder zur Behandlung einer nicht-chirurgischen Blutung sollte die Dosis wie folgt berechnet werden:
Dosis (g) = (Zielspiegel (g/l) - Basisspiegel (g/l)) x 0,043 x Körpergewicht (kg), wobei 0,043 1/Recovery ((g/l)/(g/kg)) entspricht.
In Notfällen, in denen der Fibrinogen-Basisspiegel nicht bekannt ist, wird die intravenöse Verabreichung einer initialen Dosis von 0,05 g pro kg Körpergewicht empfohlen.
Die nachfolgende Dosierung (Injektionsdosen und -häufigkeit) sollte auf Grundlage des klinischen Zustands des Patienten und der Laborergebnisse erfolgen.
Die biologische Halbwertszeit von Fibrinogen beträgt 3-4 Tage. Daher ist bei fehlendem Verbrauch eine wiederholte Behandlung mit humanem Fibrinogen in der Regel nicht erforderlich. Aufgrund der Akkumulation bei wiederholter Verabreichung zur Prophylaxe sollten die Dosis und Häufigkeit auf Grundlage der therapeutischen Ziele des Arztes für den jeweiligen Patienten bestimmt werden.
Kinder und Jugendliche
Zurzeit vorliegende Daten werden in Abschnitt 4.8 und 5.1 beschrieben; eine Dosierungsempfehlung kann jedoch nicht gegeben werden.
Art der Anwendung Intravenöse Infusion oder Injektion.
FibCLOT soll als langsame intravenöse Infusion mit einer maximalen Injektionsrate von 4 ml/min.verabreicht werden.
Hinweise zur Rekonstitution des Arzneimittels vor der Anwendung, siehe Abschnitte 6.2 und 6.6.
4.3. Gegenanzeigen
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
4.4. Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Thromboembolie
Es besteht ein Thromboserisiko, wenn Patienten mit einer besonders hohen Dosis humanen Fibrinogens oder mit wiederholten Dosen behandelt werden. Patienten, die humanes Fibrinogen erhalten, sollten engmaschig auf Anzeichen oder Symptome einer Thrombose überwacht werden.
Bei Patienten mit koronarer Herzerkrankung oder Myokardinfarkt in der Anamnese, Patienten mit Lebererkrankung, peri- oder postoperativen Patienten, Neugeborenen oder Patienten mit einem Risiko thromboembolischer Ereignisse oder disseminierter intravaskulärer Gerinnung muss der potenzielle Nutzen einer Behandlung mit Fibrinogen aus humanem Blutplasma gegen das Risiko thromboembolischer Komplikationen abgewogen werden. Vorsicht und eine engmaschige Überwachung sind geboten.
Allergische Reaktion oder anaphylaktoide Reaktionen
Wenn allergische oder anaphylaktoide Reaktionen auftreten, muss die Injektion/Infusion sofort gestoppt werden. Im Falle eines anaphylaktischen Schocks muss die standardmäßige medizinische Behandlung durchgeführt werden.
Übertragbare Erreger
Standardmaßnahmen zur Vorbeugung von Infektionen, die sich durch den Einsatz von Arzneimitteln ergeben, die aus Blut oder Blutplasma hergestellt sind, schließen die Auswahl der Spender und das Screening der einzelnen Spenden und Plasmapools auf spezifische Infektionsmarker sowie effektive Schritte zur Inaktivierung/Entfernung von Viren im Herstellungsverfahren ein. Dennoch kann bei der Verabreichung von Arzneimitteln aus menschlichem Blut oder Blutplasma die Möglichkeit der Übertragung von Krankheitserregern nicht völlig ausgeschlossen werden. Dasselbe gilt auch für bislang unbekannte oder neu aufgetretene Viren oder andere Pathogene.
Die durchgeführten Maßnahmen werden für behüllte Viren wie das Human-Immunodeficiency-Virus (HIV), Hepatitis B (HBV)- und Hepatitis C (HCV)-Virus und das unbehüllte Hepatitis A (HAV)-Virus als wirksam erachtet. Die durchgeführten Maßnahmen sind bei unbehüllten Viren wie dem Parvovirus B19 möglicherweise von begrenztem Wert. Parvovirus B19-Infektionen können für schwangere Frauen (Infektion des Feten) und für Personen mit Immundefekt oder verstärkter Erythropoese (z. B. hämolytische Anämie) schwerwiegend sein.
Bei Patienten, die regelmäßig/wiederholt mit aus humanem Plasma gewonnenem Fibrinogen behandelt werden, sollte eine entsprechende Impfung (Hepatitis A und B) erwogen werden.
Es wird dringend empfohlen, bei jeder Verabreichung von FibCLOT Name und Chargennummer des Produktes zu dokumentieren, um einen Zusammenhang zwischen Patient und Produktcharge herzustellen.
Immunogenität
Bei Substitutionstherapien mit Gerinnungsfaktoren bei anderen kongenitalen Mangelerscheinungen wurden Antikörperreaktionen beobachtet, für Fibrinogen liegen aber derzeit keine Daten vor.
Natriumspiegel
Das Produkt enthält maximal 3 mmol (oder 69 mg) Natrium/Durchstechflasche. Dies sollte bei Patienten beachtet werden, die eine strenge salzarme Diät einhalten.
Kinder und Jugendliche
Die gleichenWarnhinweise und Vorsichtsmaßnahmen beziehen sich auf Kinder und Jugendliche.
4.5. Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Es sind keine Wechselwirkungen zwischen Produkten mit humanem Fibrinogen und anderen Arzneimitteln bekannt.
4.6. Fertilität, Schwangerschaft und Stillzeit
Die Sicherheit von Fibrinogenprodukten aus humanem Blutplasma bei Anwendung während der Schwangerschaft und Stillzeit wurden nicht in kontrollierten klinischen Studien untersucht.
Die klinische Erfahrung mit Fibrinogenprodukten bei der Behandlung geburtshilflicher Komplikationen lässt keine schädlichen Wirkungen auf den Verlauf der Schwangerschaft oder die Gesundheit des Fötus oder Neugeborenen erwarten.
4.7. Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
FibCLOT hat keinen Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
4.8. Nebenwirkungen
Tabellarische Liste der Nebenwirkungen
Die in der nachstehenden Tabelle aufgeführten Nebenwirkungen wurden bei 35 Patienten mit kongenitalem Fibrinogenmangel beobachtet, die in zwei klinischen interventionellen Studien und in einer nicht-interventionellen Unbedenklichkeitsstudie eingeschlossen waren. Im Verlauf dieser Studien wurden 36 Nebenwirkungen bei 13/35 (37,1 %) Patienten gemeldet, die insgesamt 572 Infusionen FibCLOT erhalten hatten.
Die wichtigsten Nebenwirkungen sind entsprechend der MedDRA-Systemorganklassifizierung (SOC und bevorzugter Begriff) aufgeführt. Die Häufigkeit wurde pro Infusion entsprechend der folgenden Kriterien angegeben: sehr häufig (>1/10); häufig (>1/100, <1/10); gelegentlich (>1/1.000, <1/100); selten (>1/10.000, <1/1.000); sehr selten (<1/10.000); nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar). Innerhalb jeder Häufigkeitsgruppe werden die Nebenwirkungen nach abnehmendem Schweregrad angegeben.
|
Standardsystemorganklasse (SOC) gemäß MedDRA |
Nebenwirkungen |
Häufigkeit pro Infusion (N=572) |
|
Erkrankungen des Immunsystems |
Allergische/anaphylaktoide Reaktionen (einschließlich anaphylaktischer Schock, Blässe, Erbrechen, Husten, niedriger Blutdruck, Schüttelfrost, Urtikaria) |
Gelegentlich |
|
Erkrankungen des Nervensystems |
Kopfschmerz Schwindelgefühl |
Häufig Gelegentlich |
|
Erkrankungen des Ohrs und des Labyrinths |
Tinnitus |
Gelegentlich |
|
Gefäßerkrankungen |
Thromboembolische Episoden (einschließlich tiefe Beinvenenthrombose, oberflächliche Thrombophlebitis) (siehe Abschnitt 4.4) |
Gelegentlich |
|
Erkrankungen der Atemwege, des Brustraums und Mediastinums |
Asthma |
Gelegentlich |
|
Erkrankungen der Haut und des Unterhautzellgewebes |
Erythematöser Hautausschlag |
Gelegentlich |
|
Erythem |
Gelegentlich | |
|
Hautreizung |
Gelegentlich | |
|
Nachtschweiß |
Gelegentlich |
Wärmegefühl
Gelegentlich
Hinweise zur Sicherheit auf übertragbare Erreger, siehe Abschnitt 4.4.
Das allgemeine Sicherheitsprofil unterscheidet sich nicht bei Patienten, die mit FibCLOT in anderen klinischen Situationen behandelt wurden, die eine Fibrinogentherapie erforderten.
Kinder und Jugendliche
14 der 35 Patienten, die an der Unbedenklichkeitsstudie bei kongenitalem Fibrinogenmangel teilnahmen, waren jünger als 18 Jahre. Davon waren 10 wiederum jünger als 12 Jahre und 3 jünger als 6 Jahre.
Das allgemeine Sicherheitsprofil ist bei Erwachsenen und pädiatrischen Patienten gleich.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel, Paul-Ehrlich-Institut, Paul-EhrlichStraße 51-59, 63225 Langen, Telefon: +49 6103 77 0, Telefax: +49 6103 77 1234, Website: www.pei.de anzuzeigen.
4.9. Überdosierung
Um eine Überdosierung zu vermeiden, ist während der Fibrinogentherapie eine regelmäßige Überwachung des Spiegels im Blutplasma indiziert (siehe Abschnitt 4.2).
Im Fall einer Überdosierung ist das Risiko thromboembolischer Komplikationen erhöht.
5. PHARMAKOLOGISCHE EIGENSCHAFTEN
5.1. Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Antihämorrhagika, humanes Fibrinogen, ATC-Code: B02BB01
Humanes Fibrinogen (Gerinnungsfaktor I) wird in Anwesenheit von Thrombin, aktiviertem Gerinnungsfaktor XIII (FXIIIa) und Calciumionen in ein stabiles und elastisches dreidimensionales Fibrinnetz umgewandelt.
Die Verabreichung humanen Fibrinogens erhöht den Fibrinogenspiegel im Blutplasma und kann den Blutgerinnungsdefekt bei Patienten mit Fibrinogenmangel vorübergehend korrigieren.
In einer klinischen pharmakologischen Studie (FibCLOT-Dosis 0,06 g/kg) wurde eine Normalisierung globaler Gerinnungstests (z. B. aktivierte partielle Thromboplastinzeit [aPTT] und Prothrombinzeit [PT]) bei einem Fibrinogenspiegel von oder über 0,5 g/l erreicht und hielt für mindestens 3 Tage an.
Bei allen Studien zu kongenitalem Fibrinogenmangel wurde FibCLOT verabreicht für:
100 nicht-chirurgische Blutungsepisoden bei 18 Patienten (einschließlich 17 schweren Episoden bei 9 Patienten),
38 chirurgische Eingriffe bei 15 Patienten (einschließlich 10 schweren Episoden bei 7 Patienten).
Die Mehrzahl (92,8 %) der Ereignisse (128/138) wurde mit einer einzelnen Dosis von circa 3 g FibCLOT behandelt, was einer mittleren Dosis pro Infusion von 0,050 g/kg entspricht.
In einer weiteren Studie wurden 9 Patienten zur Langzeitprophylaxe mindestens 12 Monate lang einmal pro Woche mit einer mittleren Dosis von 0,059 g/kg behandelt.
Kinder und Jugendliche
FibCLOT wurde im Rahmen klinischer Studien an 14 Patienten unter 18 Jahren verabreicht. Die mittlere Dosis pro Infusion betrug 0,059 g/kg für die Behandlung 26 nicht-chirurgischer Blutungsepisoden oder zur Prävention übermäßiger Blutungen bei 14 chirurgischen Eingriffen.
Die Europäische Arzneimittel-Agentur hat für FibCLOT eine Zurückstellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien bei Patienten unter 12 Jahren entsprechend der Entscheidung über das pädiatrische Prüfkonzept (PIP) gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen).
5.2. Pharmakokinetische Eigenschaften
Die biologische Halbwertszeit von Fibrinogen im Blutplasma beträgt 3-4 Tage.
Das Produkt wird intravenös verabreicht und ist sofort im Blutplasma in der Konzentration der verabreichten Dosis verfügbar.
In einer pharmakologischen Studie wurden 14 Patienten 14 Tage lang untersucht. Nach Infusion einer FibCLOT-Dosis von 0,06 g/kg wurde die maximale Fibrinogenkonzentration innerhalb 1 Stunde erreicht. Die Konzentration nahm danach langsam ab, wobei nach 3 bis 4 Tagen der kritische Fibrinogenspiegel von 0,5 g/l im Blutplasma erreicht war.
|
Pharmakokinetische |
FibCLOT-Einzeldosis als IV-Infusion | |
|
Parameter |
(Geometrisches Mittel [CV%]) | |
|
Cmax (g/l) |
1.4 |
(24.4) |
|
t1/2 (h) |
69.3 |
(21.8) |
|
AUC0-» (g.h/l) |
114 |
(23.4) |
|
MRT (h) |
95.6 |
(20.7) |
|
Cl (ml/h/kg) |
0.53 |
(22.0) |
|
Vss (ml/kg) |
50.7 |
(16.7) |
|
IR ((g/l)/(g/kg)) |
23.5 |
(23.2) |
|
R (%) |
93.6 |
(20.9) |
Cmax = maximale Konzentration (Aktivität) ti/2 = terminale Eliminationshalbwertszeit AUC = Fläche unter der Kurve MRT = mittlere Verweilzeit Cl = Clearance
Vss = Verteilungsvolumen in stabilem Zustand IR = gesteigerte Recovery R = In-vivo-Recovery CV = Variationskoeffizient
Kinder und Jugendliche
Für pädiatrische Patienten <12 Jahren sind keine pharmakokinetischen Daten verfügbar.
5.3. Präklinische Daten zur Sicherheit
Präklinische Daten, basierend auf den konventionellen Studien zur Sicherheitspharmakologie, Toxizität bei einzelnen und wiederholten Dosen sowie zur Thrombogenität, zeigen kein besonderes Risiko für den Menschen.
Aufgrund der Art des Produkts wurden keine Studien zur Karzinogenität durchgeführt. Reproduktionsstudien bei Tieren wurden nicht durchgeführt, da Fibrinogen ein natürlicher Bestandteil des menschlichen Körpers ist.
6. PHARMAZEUTISCHE ANGABEN
6.1. Liste der sonstigen Bestandteile
Pulver: Arginin-Hydrochlorid
Isoleucin
Lysinhydrochlorid
Glycin
Natriumcitrat-Dihydrat Lösungsmittel: Wasser für Injektionszwecke.
6.2. Inkompatibilitäten
Da keine Kompatibilitätsstudien durchgeführt wurden, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Für die intravenöse Verabreichung der rekonstituierten Lösung bei Raumtemperatur wird ein herkömmliches Infusionsbesteck empfohlen.
6.3. Dauer der Haltbarkeit
3 Jahre.
Die chemische und physikalische Stabilität nach Rekonstitution wurde für 24 Stunden bei 25 °C gezeigt. Aus mikrobiologischer Sicht sollte das Produkt jedoch sofort verwendet werden.
6.4. Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Nicht über 25 °C lagern.
Nicht einfrieren.
In der Originalverpackung aufbewahren, um den Inhalt vor Licht und Feuchtigkeit zu schützen. Aufbewahrungsbedingungen nach Rekonstitution des Arzneimittels, siehe Abschnitt 6.3.
6.5. Art und Inhalt des Behältnisses
Eine Packung enthält:
Pulver (1,5 g humanes Fibrinogen) in einer farblosen Durchstechflasche (Typ-I-Glas) versiegelt mit einem silikonisierten Bromobutylstopfen, einer Aluminiumkappe und einer Kunststoffscheibe. Lösungsmittel (100 ml Wasser für Injektionszwecke) in einer Durchstechflasche (Typ-II-Glas)
versiegelt mit einem Bromobutylstopfen, einer Aluminiumkappe und einer Kunststoffscheibe. Transfersystem mit einem sterilen Entlüfter.
6.6. Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung
Rekonstitution:
Beachten Sie die aktuellen Richtlinien für eine aseptische Vorgehensweise.
Bei Bedarf erhöhen Sie die Temperatur der beiden Durchstechflaschen (Pulver und Lösungsmittel) auf Umgebungstemperatur.
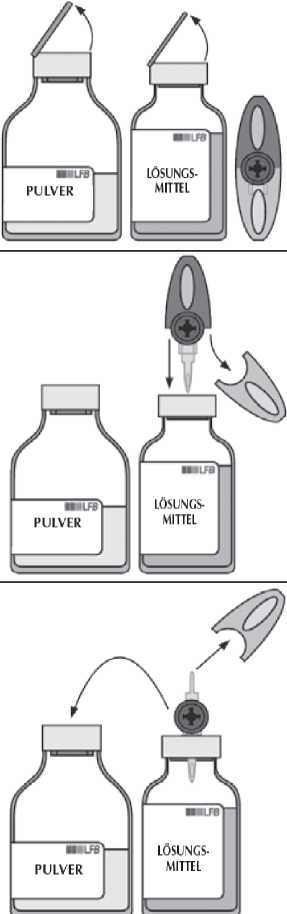
Entfernen Sie die Schutzkappen von der Durchstechflasche mit Lösungsmittel und der Durchstechflasche mit Pulver. Desinfizieren Sie die Oberfläche beider Stopfen.
Entfernen Sie die transparente Schutzhülle vom Transfersystem und stechen Sie den Einstechdorn durch die Mitte des Stopfens der Lösungsmittel-Durchstechflascheund drehen dabei gleichzeitig den Dorn hin und her.
Entfernen Sie die zweite graue Schutzhülle vom anderen Ende des Transfersystems.
Drehen Sie die Lösungsmittel-Durchstechflasche auf den Kopf und stechen Sie schnell das freiliegende Ende des Einstechdorns in die Mitte des Stopfens der Pulver-Durchstechflasche, damit das Lösungsmittel in das Pulver fließen kann.
Stellen Sie sicher, dass der Dorn immer mit Lösungsmittel bedeckt bleibt, damit das Vakuum nicht vorzeitig entlüftet wird.
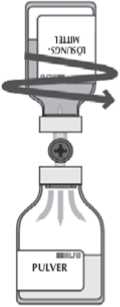
Verteilen Sie das Lösungsmittel während des Transfers durch horizontale Drehbewegungen auf der gesamten Oberfläche des Pulvers und entlang der Wand der Durchstechflasche. Stellen Sie sicher, dass das Lösungsmittel komplett überführt wurde.
Am Ende des Transfervorgangs wird das Vakuum automatisch durch sterile Luft über das Entlüftungsteil des Transfersystems entlüftet.
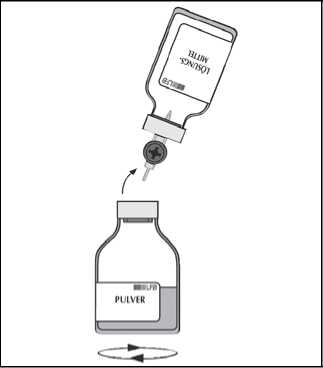
Entfernen Sie die leere Durchstechflasche (Lösungsmittel) mit dem Transfersystem.
Schwenken Sie die Lösung einige Minuten vorsichtig durch eine drehende Bewegung (damit sich kein Schaum bildet), bis das Pulver komplett gelöst ist.
Das rekonstituierte Produkt muss vor Verabreichung visuell geprüft werden, damit sichergestellt ist, dass es keine Schwebeteilchen enthält. Die rekonstituierte Lösung sollte nahezu farblos, leicht opaleszent sein. Verwenden Sie keine trüben oder mit Ablagerungen versetzte Lösungen.
Anwendung:
FibCLOT darf nur intravenös verabreicht werden, als einzelne Dosis, unmittelbar nach Rekonstitution und mit einer Geschwindigkeit von höchstens 4 ml/min.
Wenn die rekonstituierte Lösung nicht unmittelbar verabreicht wird, darf sie nicht länger als 24 Stunden bei Raumtemperatur (maximal 25 °C) gelagert werden.
Es wird empfohlen, ein Infusionsbesteck mit einem nicht sterilisierenden 15-qm-Filter zu verwenden.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den lokalen Anforderungen zu beseitigen.
7. INHABER DER ZULASSUNG
Laboratoire Fran^ais du Fractionnement et des Biotechnologies
3 Avenue des Tropiques
ZA de Courtaboeuf
91940 Les Ulis
FRANKREICH
Tel.: + 33 (0)1 69 82 70 10
Fax: + 33 (0)1 69 82 19 03
8. ZUL ASSUN GSNUMMER(N)
PEI.H.11771.01.1
9. DATUM DER ERTEILUNG DER ZULASSUNG/VERLÄNGERUNG DER ZULASSUNG
Datum der Erteilung der Zulassung:
10. STAND DER INFORMATION
Februar 2016
11. VERKAUFSABGRENZUNG
Verschreibungspflichtig
12. PLASMAHERKUNFTSLÄNDER
Deutschland, Österreich, Tschechische Republik und USA
10