Fulvestrant 1 A Pharma 250 Mg Injektionslösung In Einer Fertigspritze
Wortlaut der für die Fachinformation vorgesehenen Angaben
FACHINFORMATION
1. BEZEICHNUNG DES ARZNEIMITTELS
Fulvestrant - 1 A Pharma
250 mg Injektionslösung in einer Fertigspritze
2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
Eine Fertigspritze enthält 250 mg Fulvestrant in 5 ml Lösung.
Sonstige Bestandteile mit bekannter Wirkung:
• Ethanol (100 mg/ml)
• Benzylalkohol (100 mg/ml)
• Benzylbenzoat (150 mg/ml)
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
3. DARREICHUNGSFORM
Injektionslösung in einer Fertigspritze Klare, farblose bis gelbe, viskose Lösung.
4. KLINISCHE ANGABEN
4.1 Anwendungsgebiete
Fulvestrant - 1 A Pharma ist angezeigt zur Behandlung von postmenopausalen Frauen mit Östrogenrezeptor-positivem lokal fortgeschrittenem oder metastasiertem Mammakarzinom bei Rezidiv während oder nach adjuvanter Antiöstrogen-Therapie oder bei Progression der Erkrankung unter der Behandlung mit einem Antiöstrogen.
4.2 Dosierung und Art der Anwendung Dosierung
Erwachsene Frauen (einschließlich älterer Frauen)
Die empfohlene Dosis beträgt 500 mg in Abständen von 1 Monat, wobei 2 Wochen nach der Anfangsdosis eine zusätzliche 500 mg Dosis gegeben wird.
Besondere Patientengruppen
Niereninsuffizienz
Bei Patientinnen mit leichter bis mittelschwerer Einschränkung der Nierenfunktion (Kreatinin-Clearance > 30 ml/min) wird keine Dosisanpassung empfohlen. Die Sicherheit und Wirksamkeit bei Patientinnen mit schwerer Einschränkung der Nierenfunktion (Kreatinin-Clearance < 30 ml/min) sind nicht untersucht worden, daher ist Fulvestrant - 1 A Pharma bei diesen Patientinnen mit Vorsicht anzuwenden (siehe Abschnitt 4.4).
Leberinsuffizienz
Eine Dosisanpassung wird bei Patientinnen mit leichter bis mittelschwerer Einschränkung der Leberfunktion nicht empfohlen. Trotzdem sollte bei diesen Patientinnen Fulvestrant -1 A Pharma mit Vorsicht angewendet werden, da die Exposition von Fulvestrant erhöht sein kann. Es liegen keine Daten von Patientinnen mit schweren Leberfunktionsstörungen vor (siehe Abschnitte 4.3, 4.4 und 5.2).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Fulvestrant - 1 A Pharma bei Kindern von Geburt bis zum Alter von 18 Jahren ist nicht erwiesen. Zurzeit vorliegende Daten sind in den Abschnitten 5.1 und 5.2 beschrieben; eine Dosierungsempfehlung kann jedoch nicht gegeben werden.
Art der Anwendung
Fulvestrant - 1 A Pharma sollte langsam in Form von 2 unmittelbar aufeinander folgenden 5 ml Injektionen intramuskulär ins Gesäß appliziert werden (1-2 Minuten/Injektion), eine in jede Gesäßhälfte.
Für detailierte Hinweise zur Anwendung siehe Abschnitt 6.6.
4.3 Gegenanzeigen
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Schwangerschaft und Stillzeit (siehe Abschnitt 4.6).
Schwere Einschränkung der Leberfunktion (siehe Abschnitte 4.4 und 5.2).
4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Fulvestrant - 1 A Pharma sollte bei Patientinnen mit leichter bis mittelschwerer Einschränkung der Leberfunktion mit Vorsicht angewendet werden (siehe Abschnitte 4.2, 4.3 und 5.2).
Fulvestrant - 1 A Pharma sollte bei Patientinnen mit schwerer Einschränkung der Nierenfunktion (Kreatinin-Clearance von weniger als 30 ml/min) mit Vorsicht angewendet werden.
Aufgrund der intramuskulären Applikation sollte Fulvestrant - 1 A Pharma bei Patientinnen mit Blutungsneigung, Thrombozytopenie oder bei Patientinnen, die Antikoagulanzien erhalten, nur mit Vorsicht angewendet werden.
Thromboembolische Ereignisse werden bei Frauen mit fortgeschrittenem Brustkrebs häufig beobachtet und wurden auch in klinischen Studien mit Fulvestrant beobachtet (siehe Abschnitt 4.8). Dies sollte berücksichtigt werden, wenn Fulvestrant - 1 A Pharma für Risikopatientinnen verschrieben wird.
Es liegen keine Daten zur Langzeitwirkung von Fulvestrant auf die Knochen vor. Aufgrund des Wirkungsmechanismus von Fulvestrant besteht ein potenzielles Osteoporoserisiko.
Kinder und Jugendliche
Fulvestrant - 1 A Pharma wird für die Anwendung bei Kindern und Jugendlichen nicht empfohlen, da Sicherheit und Wirksamkeit bei dieser Patientengruppe nicht erwiesen sind (siehe Abschnitt 5.1).
Die Anwendung von Fulvestrant - 1 A Pharma kann bei Dopingkontrollen zu positiven Ergebnissen führen. Eine missbräuchliche Anwendung des Arzneimittels Fulvestrant - 1 A Pharma zu Dopingzwecken kann die Gesundheit gefährden.
Sonstige Bestandteile
Dieses Arzneimittel enthält 10 % Gewicht/Volumen (w/v) Ethanol (Alkohol). Das sind bis zu 1.000 mg pro Dosis, entsprechend 20 ml Bier oder 8 ml Wein pro Dosis.
Ein gesundheitliches Risiko besteht u. a. bei Leberkranken, Alkoholkranken,
Epileptikern, Patienten mit organischen Erkrankungen des Gehirns, Schwangeren, Stillenden und Kindern.
Dieses Arzneimittel enthält 100 mg Benyzylalkohol pro ml. Darf nicht bei Frühgeborenen oder Neugeborenen angewendet werden. Benzylalkohol kann bei Säuglingen und Kindern bis zu 3 Jahren toxische und allergische Reaktionen hervorrufen.
Dieses Arzneimittel enthält 150 mg Benzylbenzoat pro ml. Bei Neugeborenen besteht wegen des Gehalts an Benzylbenzoat ein erhöhtes Risiko für das Auftreten von Gelbsucht.
4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Eine klinische Studie zur Erfassung von Wechselwirkungen mit Midazolam (Substrat von CYP3A4) zeigte, dass Fulvestrant CYP3A4 nicht inhibiert. Klinische Interaktionsstudien mit Rifampicin (Induktor von CYP3A4) und Ketoconazol (Inhibitor von CYP3A4) zeigten keine klinisch relevanten Veränderungen in der Clearance von Fulvestrant. Daher ist eine Dosierungsanpassung für Patientinnen, die gleichzeitig Fulvestrant und CYP3A4-Inhibitoren oder -Induktoren erhalten, nicht erforderlich.
4.6 Fertilität, Schwangerschaft und Stillzeit Frauen im gebärfähigen Alter
Patientinnen im gebärfähigen Alter sollten angewiesen werden, während der Behandlung eine zuverlässige Verhütungsmethode anzuwenden.
Schwangerschaft
Fulvestrant - 1 A Pharma ist während der Schwangerschaft kontraindiziert (siehe Abschnitt 4.3). Bei Ratten und Kaninchen wurde gezeigt, dass Fulvestrant nach intramuskulären Einzeldosen die Plazenta passiert. Tierstudien haben eine Reproduktionstoxizität, einschließlich einer höheren Inzidenz von fetalen Anomalien und Todesfällen, gezeigt (siehe Abschnitt 5.3). Falls unter der Behandlung mit Fulvestrant - 1
A Pharma eine Schwangerschaft eintritt, muss die Patientin über die mögliche Gefahr für den Fetus und das potenzielle Risiko einer Fehlgeburt informiert werden.
Stillzeit
Während der Behandlung mit Fulvestrant - 1 A Pharma muss mit dem Stillen aufgehört werden. Fulvestrant geht bei laktierenden Ratten in die Muttermilch über. Es ist nicht bekannt, ob Fulvestrant beim Menschen in die Muttermilch übergeht. Aufgrund möglicher schwerwiegender Nebenwirkungen durch Fulvestrant bei Säuglingen ist die Anwendung während der Stillzeit kontraindiziert (siehe Abschnitt 4.3).
Fertilität
Beim Menschen wurden die Auswirkungen von Fulvestrant auf die Fertilität nicht untersucht.
4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Fulvestrant - 1 A Pharma hat keinen oder einen zu vernachlässigenden Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen. Während der Behandlung mit Fulvestrant - 1 A Pharma wurde jedoch sehr häufig über Asthenie berichtet. Daher ist bei Patientinnen, bei denen diese Nebenwirkung auftritt, beim Führen von Fahrzeugen und beim Bedienen von Maschinen Vorsicht geboten.
4.8 Nebenwirkungen
Dieser Abschnitt beinhaltet Informationen, die auf allen Nebenwirkungen aus klinischen Studien, Studien nach Produkteinführung oder spontanen Fallberichten basieren. Die am häufigsten angegebenen Nebenwirkungen sind Reaktionen an der Injektionsstelle, Asthenie, Übelkeit und erhöhte Leberenzymwerte (ALT, AST, ALP).
Die folgenden Häufigkeitskategorien für Nebenwirkungen wurden auf der Basis der Fulvestrant 500 mg Behandlungsgruppe in Studien-übergreifenden Sicherheitsanalysen folgender Studien berechnet: CONFIRM (Studie D6997C00002), FINDER 1 (Studie D6997C00004), FINDER 2 (Studie D6997C00006) und NEWEST (Studie D6997C00003). In diesen Studien wurde Fulvestrant 500 mg mit Fulvestrant 250 mg verglichen. Die Häufigkeitsangaben in der nachfolgenden Tabelle beruhen auf allen berichteten Ereignissen, unabhängig von der Bewertung des Kausalzusammenhangs durch den Prüfarzt.
Die unten aufgelisteten Nebenwirkungen sind entsprechend ihrer Häufigkeit und der Systemorganklasse klassifiziert. Die Häufigkeitsgruppierungen sind gemäß folgender Konvention definiert: sehr häufig (> 1/10), häufig (> 1/100, < 1/10), gelegentlich (> 1/1.000, < 1/100). Innerhalb jeder Häufigkeitsgruppierung sind die Nebenwirkungen nach abnehmendem Schweregrad geordnet.
Tabelle 1 Nebenwirkungen
|
Nebenwirkungen nach Systemorganklassen und Häufigkeit | ||
|
Infektionen und parasitäre Erkrankungen |
Häufig |
Infektionen des Harntrakts |
|
Erkrankungen des Blutes und des Lymphsystems |
Gelegentlich |
Thrombozytopenie |
|
Erkrankungen des Immunsystems |
Häufig |
Überempfindlichkeitsreaktionen |
|
Stoffwechsel- und Ernährungsstörungen |
Häufig |
Anorexiea |
|
Erkrankungen des Nervensystems |
Häufig |
Kopfschmerzen |
|
Gefäßerkrankungen |
Häufig |
Venöse Thromboemboliena, Hitzewallungen |
|
Erkrankungen des Gastrointestinaltrakts |
Sehr häufig |
Übelkeit |
|
Häufig |
Erbrechen, Durchfall | |
|
Leber- und Gallenerkrankungen |
Sehr häufig |
Erhöhte Leberenzymwerte (ALT, AST, ALP)a |
|
Häufig |
Erhöhte Bilirubinwertea | |
|
Gelegentlich |
Leberversagenc, Hepatitisc, erhöhte Gamma-GT -Werte | |
|
Erkrankungen der Haut und des Unterhautzellgewebes |
Häufig |
Hautausschlag |
|
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen |
Häufig |
Rückenschmerzena |
|
Erkrankungen der Geschlechtsorgane und der Brustdrüse |
Gelegentlich |
Vaginale Candidose, Leukorrhö, vaginale Blutungen |
|
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort |
Sehr häufig |
Astheniea, Reaktionen an der Injektionsstelleb |
|
Gelegentlich |
Blutungen an der Injektionsstelle, Hämatome an der Injektionsstelle |
a Schließt Nebenwirkungen ein, für die infolge der Grunderkrankung das genaue Ausmaß des Beitrags von Fulvestrant nicht bewertet werden kann. b Der Begriff Reaktionen an der Injektionsstelle umfasst nicht die Begriffe Blutungen an der Injektionsstelle und Hämatome an der Injektionsstelle. c Das Ereignis wurde nicht im Rahmen der großen klinischen Studien (CONFIRM, FINDER 1, FINDER 2, NEWEST) beobachtet. Die Häufigkeit wurde berechnet, in dem der obere Grenzwert des 95 %-Konfidenzintervalls als Punktschätzwert herangezogen wurde. Dies wird berechnet mit 3/563 (wobei 563 die Anzahl an Patientinnen in den großen klinischen Studien darstellt), was der Häufigkeitskategorie „gelegentlich“ entspricht.
Selten können Überempfindlichkeitsreaktionen durch Benzylalkohol auftreten.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-RisikoVerhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem
Bundesinstitut für Arzneimittel und Medizinprodukte Abt. Pharmakovigilanz
Kurt-Georg-Kiesinger-Allee 3 D-53175 Bonn Website: www.bfarm.de
anzuzeigen.
4.9 Überdosierung
Beim Menschen liegen keine Erfahrungen mit einer Überdosierung vor. Tierstudien weisen darauf hin, dass sich unter höheren Fulvestrantdosen keine anderen Wirkungen zeigen als solche, die direkt oder indirekt in Zusammenhang mit der antiöstrogenen Wirkung stehen (siehe Abschnitt 5.3). Falls eine Überdosierung eintritt, wird eine unterstützende symptomatische Behandlung empfohlen.
5. PHARMAKOLOGISCHE EIGENSCHAFTEN
5.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Endokrine Therapie, Antiestrogene ATC-Code: L02BA03
Wirkmechanismus und pharmakodynamische Wirkungen
Fulvestrant ist ein kompetitiver Östrogenrezeptor (ER)-Antagonist mit einer dem Östradiol vergleichbaren Affinität. Fulvestrant blockiert die trophischen Wirkungen der Östrogene, ohne partiell agonistische (östrogenartige) Aktivität. Der Wirkungsmechanismus ist mit einer Reduktion der Östrogenrezeptorprotein-Spiegel verknüpft.
Klinische Studien mit postmenopausalen Frauen mit primärem Mammakarzinom haben gezeigt, dass Fulvestrant das ER-Protein in ER-positiven Tumoren im Vergleich zu Placebo signifikant reduziert. In Übereinstimmung mit dem Fehlen von intrinsischen Östrogen-agonistischen Wirkungen war die Expression des Progesteronrezeptors ebenfalls signifikant vermindert. Es wurde auch gezeigt, dass Fulvestrant 500 mg bei Mammakarzinomen unter postmenopausalen, neoadjuvanten Bedingungen den Östrogenrezeptor und den Proliferationsmarker Ki67 stärker reduziert als Fulvestrant 250 mg.
Klinische Sicherheit und Wirksamkeit bei fortgeschrittenem Mammakarzinom
Es wurde 1 klinische Phase-III-Studie mit 736 postmenopausalen Frauen mit fortgeschrittenem Mammakarzinom durchgeführt, die ein Wiederauftreten der Erkrankung während oder nach einer adjuvanten Hormontherapie oder in der Folge der Hormontherapie der fortgeschrittenen Erkrankung eine Progression zeigten. Die Studie umfasste 423 Patientinnen, deren Erkrankung während einer Antiöstrogen-Therapie wieder aufgetreten oder fortgeschritten war (AE Untergruppe) und 313 Patientinnen, deren Erkrankung während einer Aromatasehemmer-Therapie wieder aufgetreten oder fortgeschritten war (AI Untergruppe). In dieser Studie wurde die Wirksamkeit und Sicherheit von Fulvestrant 500 mg (n = 362) mit Fulvestrant 250 mg (n = 374) verglichen. Das Progressions-freie Überleben (Progression-free survival, PFS) war der primäre Endpunkt; entscheidende sekundäre Endpunkte zur Wirksamkeit waren objektive Ansprechrate (Objective response rate, ORR), klinische Nutzenrate (Clinical benefit rate, CBR) und Gesamtüberleben (Overall survival, OS). Ergebnisse zur Wirksamkeit aus der CONFIRM-Studie sind in Tabelle 2 zusammengefasst.
Tabelle 2 Zusammenfassung der Ergebnisse für den primären Endpunkt zur Wirksamkeit (PFS) und entscheidende sekundäre Endpunkte zur Wirksamkeit aus der CONFIRM-Studie
|
Variable |
Art der Schätzung; Vergleich der Behandlung |
Fulvestrant 500 mg (n = 362) |
Fulvestrant 250 mg (n = 374) |
Vergleich zwischen Gruppen (Fulvestrant 500 mg/Fulvestrant 250 mg) | ||
|
Hazard Ratio |
95 %-CI |
p-Wert | ||||
|
PFS |
K-M-Median in Monaten; Hazard Ratio | |||||
|
Alle Patientinnen |
6,5 |
5,5 |
0,80 |
0,68; 0,94 |
0,006 | |
|
- AE Untergruppe (n = 423) |
8,6 |
5,8 |
0,76 |
0,62; 0,94 |
0,013 | |
|
- AI Untergruppe (n = 313)a |
5,4 |
4,1 |
0,85 |
0,67; 1,08 |
0,195 | |
|
OSb |
K-M-Median in Monaten; Hazard Ratio | |||||
|
Alle Patientinnen |
26,4 |
22,3 |
0,81 |
0,69; 0,96 |
0,016c | |
|
- AE Untergruppe (n = 423) |
30,6 |
23,9 |
0,79 |
0,63; 0,99 |
0,038c | |
|
- AI Untergruppe (n = 313)a |
24,1 |
20,8 |
0,86 |
0,67; 1,11 |
0,241c | |
|
Variable |
Art der Schätzung; Vergleich der Behandlung |
Fulvestrant 500 mg (n = 362) |
Fulvestrant 250 mg (n = 374) |
Vergleich zwischen Gruppen (Fulvestrant 500 mg/Fulvestrant 250 mg) | ||
|
Absoluter Unterschied in % |
95 %-CI | |||||
|
ORRd |
% der Patientinnen mit OR; absoluter Unterschied in % | |||||
|
Alle Patientinnen |
13,8 |
14,6 |
-0,8 |
-5,8; 6,3 | ||
|
- AE Untergruppe (n = 296) |
18,1 |
19,1 |
-1,0 |
-8,2; 9,3 | ||
|
- AI Untergruppe (n = 205)a |
7,3 |
8,3 |
-1,0 |
-5,5; 9,8 | ||
|
CBRe |
% der Patientinnen mit CB; absoluter Unterschied in % | |||||
|
Alle Patientinnen |
45,6 |
39,6 |
6,0 |
-1,1; 13,3 | ||
|
- AE Untergruppe (n = 423) |
52,4 |
45,1 |
7,3 |
-2,2; 16,6 | ||
|
- AI Untergruppe (n = 313)a |
36,2 |
32,3 |
3,9 |
-6,1; 15,2 | ||
a Fulvestrant ist angezeigt für Patientinnen, deren Erkrankung während einer Antiöstrogen-Therapie wieder aufgetreten oder fortgeschritten war. Die Ergebnisse in der AI Untergruppe sind nicht beweiskräftig. b Die Darstellung des OS bezieht sich auf die finalen Überlebensanalysen bei einer Datenreife von 75 %.
c Nominaler p-Wert ohne Multiplizitätsbereinigung zwischen den initialen Gesamtüberlebensanalysen bei einer Datenreife von 50 % und den aktualisierten Überlebensanalysen bei einer Datenreife von 75 %. d ORR wurde bei den Patientinnen beurteilt, die bezüglich des Ansprechens bei Studienbeginn auswertbar waren (d. h. die mit einer messbaren Erkrankung bei Studienbeginn: 240 Patientinnen in der Fulvestrant 500 mg Gruppe und 261 Patientinnen in der Fulvestrant 250 mg Gruppe). e Patientinnen mit bestem objektivem Ansprechen in Form vollständigen Ansprechens, teilweisen Ansprechens oder stabiler Erkrankung > 24 Wochen.
PFS: Progression-free survival (Progressions-freies Überleben); ORR: Objective response rate (objektive Ansprechrate); OR: Objective response (objektives Ansprechen); CBR: Clinical benefit rate (klinische Nutzenrate); CB: Clinical benefit (klinischer Nutzen); OS: Overall survival (Gesamtüberleben); K-M: Kaplan-Meier; CI: Confidence intervall (Konfidenzintervall); AI: Aromatase inhibitor (Aromatasehemmer); AE: Anti-estrogen (Antiöstrogen).
Es wurden 2 klinische Phase-III-Studien mit insgesamt 851 postmenopausalen Frauen mit fortgeschrittenem Mammakarzinom durchgeführt, die ein Wiederauftreten der Erkrankung während oder nach einer adjuvanten Hormontherapie oder in der Folge der Hormontherapie der fortgeschrittenen Erkrankung eine Progression zeigten. Im Patientinnenkollektiv der Studien hatten 77 % Östrogenrezeptor-positiven Brustkrebs. In diesen Studien wurden die Sicherheit und Wirksamkeit einer monatlichen Anwendung von Fulvestrant 250 mg mit der täglichen Einnahme von 1 mg Anastrozol (Aromatasehemmer) verglichen. Insgesamt war Fulvestrant in einer einmal monatlichen Dosierung von 250 mg im Hinblick auf das Progressions-freie Überleben, das objektive Ansprechen und Zeit bis zum Tod mindestens ebenso wirksam wie Anastrozol. Für keinen dieser Endpunkte gab es statistisch signifikante Unterschiede zwischen den beiden Behandlungsgruppen. Das Progressions-freie Überleben war der primäre Endpunkt. Die gemeinsame Auswertung beider Studien hat gezeigt, dass es bei 83 % der Patientinnen, die Fulvestrant erhielten, zu einer Progression kam, verglichen mit 85 % der Patientinnen, die Anastrozol erhielten. Die gemeinsame Auswertung beider Studien hat gezeigt, dass die Hazard Ratio von Fulvestrant 250 mg gegenüber Anastrozol für das Progressions-freie Überleben 0,95 (95 % CI 0,82-1,10) war. Die objektive Ansprechrate für Fulvestrant 250 mg betrug 19,2 % im Vergleich zu 16,5 % für Anastrozol. Die mediane Zeit bis zum Tod betrug 27,4 Monate für die mit Fulvestrant behandelten Patientinnen und 27,6 Monate für Patientinnen, die mit Anastrozol behandelt wurden. Die Hazard Ratio von Fulvestrant 250 mg gegenüber Anastrozol für die Zeit bis zum Tod war 1,01 (95 % CI 0,86-1,19).
Wirkungen auf das postmenopausale Endometrium
Präklinische Daten deuten nicht darauf hin, dass Fulvestrant eine stimulierende Wirkung auf das postmenopausale Endometrium hat (siehe Abschnitt 5.3). Eine Studie über 2 Wochen mit postmenopausalen gesunden Probandinnen, die mit 20 Mikrogramm Ethinylestradiol pro Tag behandelt wurden, hat gezeigt, dass eine Vorbehandlung mit Fulvestrant 250 mg im Vergleich zur Vorbehandlung mit Placebo zu einer signifikant verminderten Stimulierung des postmenopausalen Endometriums führte. Das wurde durch Ultraschallmessungen der Dicke des Endometriums ermittelt.
Eine neoadjuvante Behandlung mit einer Dauer von bis zu 16 Wochen führte bei Patientinnen mit Mammakarzinom, die entweder mit Fulvestrant 500 mg oder Fulvestrant 250 mg behandelt wurden, nicht zu klinisch signifikanten Änderungen der Endometriumdicke. Dies weist auf das Fehlen eines agonistischen Effekts hin. Es liegen keine Anzeichen für Nebenwirkungen auf das Endometrium bei den untersuchten Patientinnen mit Mammakarzinom vor. Bezüglich der Morphologie des Endometriums sind keine Daten verfügbar.
In 2 Kurzzeit-Studien (1 und 12 Wochen) mit prämenopausalen Patientinnen mit gutartiger gynäkologischer Erkrankung wurden zwischen Fulvestrant- und PlaceboGruppen keine signifikanten Unterschiede in der Dicke des Endometriums (gemessen mit Ultraschall) beobachtet.
Wirkungen auf die Knochen
Es gibt keine Langzeitdaten zur Wirkung von Fulvestrant auf die Knochen. Eine neoadjuvante Behandlung mit einer Dauer von bis zu 16 Wochen führte bei Patientinnen mit Mammakarzinom, die entweder mit Fulvestrant 500 mg oder Fulvestrant 250 mg behandelt wurden, zu keinen klinisch signifikanten Änderungen von Markern für Knochenabbau im Serum.
Kinder und Jugendliche
Fulvestrant ist für die Anwendung bei Kindern nicht indiziert. Die Europäische Arzneimittel-Agentur hat für Fulvestrant eine Freistellung von der Verpflichtung zur Vorlage von Ergebnissen aus Studien in allen pädiatrischen Altersklassen im Anwendungsgebiet Mammakarzinom gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen).
Bei 30 Mädchen im Alter von 1-8 Jahre mit progressiver vorzeitiger Pubertät im Zusammenhang mit McCune-Albright-Syndrom (MAS) wurden in 1 offenen Phase-II-Studie die Sicherheit, Wirksamkeit und Pharmakokinetik von Fulvestrant untersucht. Die pädiatrischen Patienten erhielten monatlich eine intramuskuläre Fulvestrant-Dosis von 4 mg/kg. Diese 12-monatige Studie untersuchte eine Anzahl von MAS-Endpunkten und zeigte eine Reduktion in der Häufigkeit von Vaginalblutungen und eine Reduktion in der Rate fortschreitender Knochenalterung. Die Steady-State-Konzentrationen (trough level) von Fulvestrant bei den Kindern in dieser Studie waren konsistent mit denjenigen bei Erwachsenen (siehe Abschnitt 5.2). Aus dieser kleinen Studie haben sich keine neuen Sicherheitsbedenken ergeben, 5 Jahres-Daten sind allerdings noch nicht verfügbar.
5.2 Pharmakokinetische Eigenschaften Resorption
Nach Anwendung von Fulvestrant, das nach intramuskulärer Injektion lang wirksam ist, wird Fulvestrant langsam resorbiert und maximale Plasmakonzentrationen (Cmax) werden nach ungefähr 5 Tagen erreicht. Bei der Anwendung des Fulvestrant 500 mg Dosierungsregimes werden innerhalb des 1. Monats nach der Anwendung Expositionslevel auf, oder annähernd auf, dem Niveau des Steady State erreicht (mittlerer [CV]: AUC 475 [33,4 %] ngTage/ml, Cmax 25,1 [35,1 %] ng/ml, bzw. Cmm 16,3 [25.9 %] ng/ml). Im Steady State bleiben die Plasmakonzentrationen von Fulvestrant in einem relativ engen Bereich mit einer bis zu ungefähr 3-fachen Differenz zwischen maximalen und minimalen Konzentrationen. Nach intramuskulärer Applikation ist im Dosierungsbereich von 50-500 mg die Exposition annähernd zur Dosis proportional.
Verteilung
Fulvestrant unterliegt einer extensiven und schnellen Verteilung. Das große scheinbare Verteilungsvolumen im Steady State (Vdss) von ungefähr 3-5 l/kg zeigt, dass überwiegend eine extravasale Verteilung vorliegt. Fulvestrant wird in hohem Maße an Plasmaproteine gebunden (99 %). Fraktionen von Lipoprotein sehr niedriger Dichte (VLDL), Lipoprotein niedriger Dichte (LDL) und Lipoprotein hoher Dichte (HDL) sind die hauptsächlichen Bindungspartner. Interaktionsstudien zur kompetitiven Proteinbindung wurden nicht durchgeführt. Die Rolle des Geschlechtshormon-bindenden Globulins (SHBG) wurde nicht untersucht.
Biotransformation
Der Metabolismus von Fulvestrant ist nicht vollständig geklärt, beinhaltet aber Kombinationen einer Anzahl möglicher Biotransformationswege, die denen endogener Steroide entsprechen. Die identifizierten Metaboliten (einschließlich 17-Keton-, Sulfon-, 3-Sulfat-, 3- und 17-Glucuronidmetaboliten) sind in Antiöstrogenmodellen entweder weniger wirksam oder zeigen eine ähnliche Aktivität wie Fulvestrant. Studien an menschlichen Leberpräparaten und rekombinanten menschlichen Enzymen zeigen, dass CYP3A4 das einzige P450 Isoenzym ist, das an der Oxidation von Fulvestrant beteiligt ist, jedoch scheinen in vivo nicht durch P450 vermittelte Biotransformationswege zu überwiegen. In-vitro-Daten weisen darauf hin, dass Fulvestrant die CYP450-Isoenzyme nicht inhibiert.
Elimination
Fulvestrant wird hauptsächlich in metabolisierter Form eliminiert. Die Ausscheidung erfolgt hauptsächlich über die Fäzes mit weniger als 1 % Ausscheidung über den Urin. Fulvestrant hat eine hohe Clearance, 11 ± 1,7 ml/min/kg, die auf ein hohes hepatisches Extraktionsverhältnis hinweist. Die terminale Halbwertszeit (ti/2) nach intramuskulärer Applikation wird von der Absorptionsrate bestimmt und wurde auf 50 Tage berechnet.
Besondere Patientinnengruppen
In einer pharmakokinetischen Patientinnengruppen-Analyse von Daten aus Phase-III-Studien wurde im Hinblick auf Alter (Bereich von 33-89 Jahre), Gewicht (40-127 kg) oder Rasse kein Unterschied im pharmakokinetischen Profil von Fulvestrant festgestellt.
Niereninsuffizienz
Eine leichte bis mäßige Nierenfunktionsstörung hat die Pharmakokinetik von Fulvestrant in keinem klinisch relevanten Ausmaß beeinflusst.
Leberinsuffizienz
Die Pharmakokinetik von Fulvestrant wurde in klinischen Einzeldosis-Studien an Studienteilnehmern mit leichten bis mittelschweren Leberfunktionsstörungen (Child-Pugh-Klassen A und B) untersucht. Dabei wurde eine hohe Dosis einer Formulierung für eine kürzer wirksame intramuskuläre Injektion angewendet. Bei den Studienteilnehmern mit Leberfunktionsstörung zeigte sich ein 2,5-facher Anstieg des AUC-Werts im Vergleich zu den gesunden Studienteilnehmern. Bei Patientinnen, die Fulvestrant erhalten, wird erwartet, dass eine Erhöhung der Exposition dieser Größenordnung gut vertragen wird. Personen mit schweren Leberfunktionsstörungen (Child-Pugh-Klasse C) wurden nicht ausgewertet.
Kinder und Jugendliche
Die Pharmakokinetik von Fulvestrant wurde in 1 klinischen Studie evaluiert, die an 30 Mädchen mit progressiver vorzeitiger Pubertät im Zusammenhang mit McCune-Albright-Syndrom durchgeführt wurde (siehe Abschnitt 5.1). Die pädiatrischen Patienten waren im Alter von 1-8 Jahre und erhielten monatlich eine intramuskuläre Fulvestrant-Dosis von 4 mg/kg. Das geometrische Mittel (Standardabweichung) der Steady-State-Konzentration (trough level; Cminss) und AUCss war 4,2 (0,9) ng/ml bzw. 3.680 (1.020) ngh/ml. Obwohl die erhobenen Daten limitiert waren, scheinen die Steady-State-Konzentrationen (trough level) von Fulvestrant bei Kindern mit denen bei Erwachsenen konsistent zu sein.
5.3 Präklinische Daten zur Sicherheit
Die akute Toxizität von Fulvestrant ist gering.
Fulvestrant Injektionslösung und andere Formulierungen von Fulvestrant wurden in Mehrfachdosis-Studien von allen Tierspezies gut vertragen. Lokale Reaktionen, darunter Myositis und Granuloma an der Injektionsstelle, wurden auf die Trägersubstanz zurückgeführt, allerdings war bei Kaninchen der Schweregrad der Myositis unter Fulvestrant, verglichen mit der salinischen Kontrolle, höher. In Toxizitätsstudien mit intramuskulärer Mehrfachdosierung von Fulvestrant an Ratten und Hunden war die antiöstrogene Wirkung von Fulvestrant für die meisten der beobachteten Effekte verantwortlich, insbesondere beim weiblichen Fortpflanzungssystem, aber auch bei anderen hormonempfindlichen Organen beider Geschlechter. Eine Arterienentzündung, die verschiedene Gewebebereiche betraf, wurde bei einigen Hunden nach chronischer Dosierung (12 Monate) beobachtet.
In Studien an Hunden wurden nach oraler und intravenöser Anwendung Auswirkungen auf das Herz-Kreislauf-System beobachtet (leichte Erhöhung des S-T-Segments im EKG [oral] und Sinusstillstand bei einem Hund [intravenös]). Diese traten bei Expositionen auf, die höher waren als bei Patientinnen (Cmax mehr als das 15-Fache), und sind wahrscheinlich in der klinischen Dosierung für die Sicherheit des Menschen von begrenzter Bedeutung.
Fulvestrant hat kein genschädigendes Potenzial aufgewiesen.
In Dosierungen, die vergleichbar mit der klinischen Dosierung waren, beeinflusste Fulvestrant entsprechend seiner antiöstrogenen Wirkung die Fortpflanzung und die Embryonal-/Fetalentwicklung. Bei Ratten wurde eine reversible Verringerung von weiblicher Fertilität und Überlebensrate der Embryonen, Dystokie und vermehrtes Auftreten fetaler Missbildungen, einschließlich Tarsalflexuren, beobachtet. Kaninchen, die Fulvestrant erhielten, konnten die Trächtigkeit nicht aufrechterhalten. Zunahmen des Plazentagewichts und Verluste der Feten nach der Implantation wurden beobachtet. Bei Kaninchen war die Inzidenz fetaler Variationen erhöht (rückwärtige Verlagerung des Beckenhüftgürtels und 27 präsakrale Wirbel).
Eine Kanzerogenitätsstudie über 2 Jahre an Ratten (intramuskuläre Anwendung von Fulvestrant) zeigte unter der hohen Dosierung von 10 mg/Ratte/15 Tage eine erhöhte Inzidenz von gutartigen Granulosazelltumoren der Ovarien bei weiblichen und eine erhöhte Inzidenz von Leydig-Zell-Tumoren im Hoden bei männlichen Ratten. Im Rahmen einer 2-jährigen Kanzerogenitätsstudie an Mäusen (tägliche orale Einnahme) kam es bei Dosen von 150 und 500 mg/kg/Tag zu einem erhöhten Auftreten von Keimstrang-StromaTumoren des Ovars (sowohl gut- als auch bösartig). Auf dem No-effect-level für diese Befunde betrugen die systemischen Expositionslevel (AUC) bei weiblichen Ratten ungefähr das 1,5-Fache bzw. bei männlichen Ratten das 0,8-Fache der erwarteten menschlichen Expositionslevel und bei männlichen und weiblichen Mäusen ungefähr das 0,8-Fache der erwarteten menschlichen Expositionslevel. Die Induktion solcher Tumoren steht im Einklang mit pharmakologisch bedingten Veränderungen endokriner Feedbackmechanismen der Gonadotropinspiegel, die bei gebärfähigen Tieren durch Antiöstrogene hervorgerufen werden. Deshalb werden diese Befunde für die Anwendung von Fulvestrant bei postmenopausalen Frauen mit fortgeschrittenem Mammakarzinom nicht als relevant angesehen.
6. PHARMAZEUTISCHE ANGABEN 6.1 Liste der sonstigen Bestandteile
• Ethanol 96 %
• Benzylalkohol
• Benzylbenzoat
• natives Rizinusöl
6.2 Inkompatibilitäten
Da keine Kompatibilitätsstudien durchgeführt wurden, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
6.3 Dauer der Haltbarkeit 2 Jahre
6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Im Kühlschrank lagern (2°C - 8°C).
Die Fertigspritze in der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen.
6.5 Art und Inhalt des Behältnisses
Fulvestrant - 1 A Pharma ist in Packungen mit 1 oder 2 Fertigspritze/n zur Einmalanwendung erhältlich, bestehend aus einem Spritzenzylinder aus silikonisiertem Typ I Glas, der mit einem Originalitätsverschluss/Luer-Lock-Anschluss, einem silikonisierten Brombutylkolbenstopfen, einer Kappe aus Brombutyl/synthetischem Isopren, einer Polystyrolkolbenstange und einem Backstop aus Polypropylen versehen ist. Außerdem ist für jede Fertigspritze eine sterile Injektionsnadel mit Sicherheitssystem (Sicherheitsinjektionsnadel) beigefügt.
6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung
Anwendungshinweise
Warnhinweis - Die Sicherheitsinjektionsnadel darf vor der Anwendung nicht autoklaviert werden. Die Hände müssen während der Anwendung und Entsorgung immer hinter der Nadel bleiben.
Die Fertigspritzen werden mit einer BD SafetyGlide® oder Terumo SurGuard® Sicherheitsinjektionsnadel geliefert.
Anwendungshinweise für die BD SafetyGlide® Sicherheitsinjektionsnadel Bei jeder der beiden Spritzen:
• Entnehmen Sie die Spritze und die Sicherheitsinjektionsnadel vorsichtig aus der Packung.
• Entfernen Sie die Schutzkappe von der Spitze des Spritzenzylinders.
• Öffnen Sie die äußere Verpackung der Sicherheitsinjektionsnadel (BD Safety Glide®). Verbinden Sie die Injektionsnadel mit dem Luer-Lock-Anschluss.
• Schrauben Sie die Injektionsnadel auf das Luer-Lock-Verbindungsstück. Schrauben Sie so lange, bis beide Teile fest miteinander verbunden sind.
• Ziehen Sie die Schutzabdeckung gerade von der Spritze ab, um eine Beschädigung der Nadelspitze zu vermeiden.
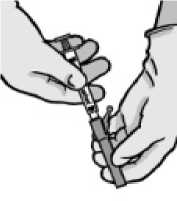
• Entfernen Sie die Injektionsnadelhülle.
Halten Sie die Spritze mit der Nadel nach oben und drücken Sie den Kolben behutsam bis sich das Arzneimittel oben in der Spritze befindet. Es sollte keine Luft mehr im Spritzenzylinder sein.
• Verabreichen Sie die Injektion langsam intramuskulär (1-2 Minuten/Injektion) in das Gesäß. Damit das Produkt möglichst anwenderfreundlich ist, liegt die Nadelöffnung oben, wenn der Hebelarm ebenfalls oben ist.
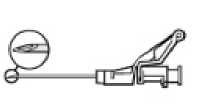
Nach Beendigung der Injektion geben Sie dem aktivierungsgesteuerten Hebelarm sofort mit einem Finger einen Stoß, um den Sicherheitsmechanismus zu aktivieren.
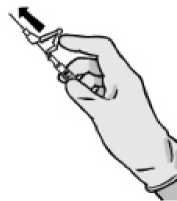
HINWEIS: Halten Sie die Spritze beim Aktivieren von sich selbst und von anderen weg. Achten Sie auf den „Klick“ und überzeugen Sie sich visuell davon, dass die Nadelspitze vollständig bedeckt ist.
Anwendungshinweise für die Terumo SurGuard® Sicherheitsinjektionsnadel
Bei jeder der beiden Spritzen:
• Entnehmen Sie die Spritze und die Sicherheitsinjektionsnadel vorsichtig aus der Packung.
• Entfernen Sie die Schutzkappe von der Spitze des Spritzenzylinders.
• Befestigen Sie die Spritze unter Verwendung einer aseptischen Arbeitsweise an der Sicherheitsinjektionsnadel. Greifen Sie dazu die Nadel an der Basis, nicht an der Schutzhülle, und drehen Sie die Spritze im Uhrzeigersinn fest.
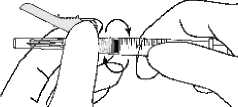
• Ziehen Sie den Nadelschutz von der Nadel weg in Richtung Spritzenzylinder bis zu dem gezeigten Winkel. Entfernen Sie dann die Injektionsnadelhülle.
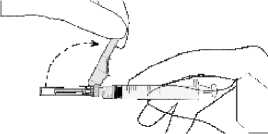
• Halten Sie die Spritze mit der Nadel nach oben und drücken Sie den Kolben behutsam bis sich das Arzneimittel oben in der Spritze befindet. Es sollte keine Luft mehr im Spritzenzylinder sein.
• Verabreichen Sie die Injektion langsam intramuskulär (1-2 Minuten/Injektion) in das Gesäß.
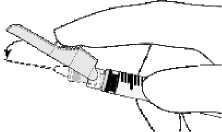
• Nach Beendigung der Injektion ziehen Sie die Nadel aus der Haut und aktivieren Sie den Schutzmechanismus einhändig durch eine der folgenden 3 Methoden:
- Aktivierung mit dem Zeigefinger
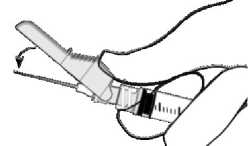
- Aktivierung mit dem Daumen
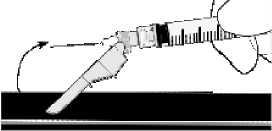
Aktivierung auf einer Oberfläche
Die Aktivierung wird durch einen hörbaren und/oder spürbaren „Klick” bestätigt und kann visuell überprüft werden. Falls Sie sich nicht sicher sind, dass der Nadelschutz vollständig aktiviert ist, wiederholen Sie diesen Schritt.
Entsorgung
Die Fertigspritzen sind ausschließlich zur einmaligen Anwendung vorgesehen.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
7. INHABER DER ZULASSUNG
1 A Pharma GmbH Keltenring 1 + 3 82041 Oberhaching Telefon: 089/6138825-0 Telefax: 089/6138825-65 E-Mail: medwiss@1apharma.com
8. ZULASSUNGSNUMMER
92985.00.00
9. DATUM DER ERTEILUNG DER ZULASSUNG
Datum der Erteilung der Zulassung: (siehe Unterschrift)
10. STAND DER INFORMATION
11. VERKAUFSABGRENZUNG
V erschreibungspflichtig