Ig Vena 50 G/L Infusionslösung
ZUSAMMENFASSUNG DER MERKMALE DES ARZNEIMITTELS
1 BEZEICHNUNG DES ARZNEIMITTELS
Ig Vena 50 g/l Infusionslösung
2 QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
Normales Immunglobulin vom Menschen (IVIg).
Ein ml Lösung enthält:
Normales Immunglobulin vom Menschen 50 mg (Reinheit von mindestens 95% IgG)
Eine Durchstechflasche von 20 ml enthält: 1 g normales Immunglobulin vom Menschen Eine Durchstechflasche von 50 ml enthält: 2,5 g normales Immunglobulin vom Menschen Eine Durchstechflasche von 100 ml enthält: 5 g normales Immunglobulin vom Menschen Eine Durchstechflasche von 200 ml enthält: 10 g normales Immunglobulin vom Menschen
Verteilung der IgG Subklassen (ungefähre Werte):
IgGx 62,1 %
IgG2 34,8 %
IgG3 2,5 %
IgG4 0,6 %
Der maximale IgA Gehalt beträgt 50 Mikrogramm/ml
Hergestellt aus Plasma von menschlichen Spendern.
Sonstige Bestandteile mit bekannter Wirkung:
Die Lösung enthält 100 mg/ ml Maltose
Dieses Arzneimittel enthält 3 mmol/Liter (oder 69 mg) Natrium. Dies soll bei Patienten, die eine kontrollierte Natriumdiät einhalten müssen, beachtet werden.
Die vollständige Auflistung der sonstigen Bestandteile siehe Abschnitt 6.1.
3 DARREICHUNGSFORM
Infusionslösung.
Die Lösung sollte klar oder leicht opaleszent und farblos oder hellgelb sein.
KLINISCHE ANGABEN
4
4.1 Anwendungsgebiete
Substitutionsbehandlung bei Erwachsenen, und Kindern und Jugendlichen (0-18 Jahre) bei:
• Primären Immunmangelkrankheiten mit eingeschränkter Antikörperproduktion (siehe Abschnitt 4.4):
• Hypogammaglobulinämie und rezidivierenden bakteriellen Infektionen in Patienten mit chronischer lymphatischer Leukämie, welche nicht auf die prophylaktische Gabe von Antibiotika angesprochen haben.
• Hypogammaglobulinämie und rezidivierenden bakteriellen Infektionen in Patienten in der Plateauphase des multiplen Myeloms, die nicht auf eine Pneumokokkenimmunisierung angesprochen haben
• Hypogammaglobulinämie in Patienten nach allogener hämatopoetische Stammzelltransplantation (HSZT).
• Kongenitalem AIDS mit rezidivierenden bakteriellen Infektionen
Immunmodulation in Erwachsenen, und Kindern und Jugendlichen (0-18 Jahre) bei:
• Primärer Immunthrombozytopenie (ITP) in Patienten mit hohem Blutungsrisiko oder auch vor Operationen zur Korrektur der Thrombozytenzahl.
• Guillain Barre Syndrom.
• Chronisch inflammatorisch demyelinisierender Polyneuropathie (CIDP).
• Kawasaki Syndrom.
4.2 Dosierung, Art und Dauer der Anwendung
Der Beginn und die Überwachung einer Substitutionstherapie sollten unter Aufsicht eines Arztes
mit Erfahrung in der Behandlung von Immundefekten stattfinden.
Dosierung
Dosierung und Dosisintervalle richten sich nach der jeweiligen Indikation.
Bei Substitutionsbehandlungen kann eine individuelle Dosierung für jeden Patienten in
Abhängigkeit von der pharmakokinetischen und klinischen Reaktion notwendig sein. Folgende
Dosierungsangaben können als Richtlinie gelten.
Substitutionsbehandlung bei primären Immundefekten:
Bei der Dosierung sollte ein IgG-Talspiegel von mindestens 5 - 6 g/l angestrebt werden (gemessen vor der nächsten Infusion). Nach Behandlungsbeginn werden 3 - 6 Monate benötigt, um ein Gleichgewicht einzustellen. Die empfohlene Initialdosis beträgt einmalig 0,4 - 0,8 g/kg Körpergewicht (KG) gefolgt von mindestens 0,2 g/kg KG alle 3 - 4 Wochen.
Um einen Talspiegel von 5 - 6 g/l aufrechtzuerhalten, ist eine Erhaltungsdosis von 0,2 - 0,8 g/kg KG pro Monat erforderlich. Die Dosierungsintervalle können bei Vorliegen eines steady states 3 - 4 Wochen betragen.
Die Talspiegel sollten regelmäßig kontrolliert und im Zusammenhang mit der Infektionshäufigkeit bewertet werden. Es kann notwendig sein die Dosierung zu erhöhen und einen höheren Talspiegel anzustreben, um die Infektionshäufigkeit zu vermindern.
Hypogammaglobulinämie und rezidivierende bakterielle Infektionen in Patienten mit chronischer lymphatischer Leukämie, welche nicht auf die prophylaktische Gabe von Antibiotika angesprochen haben; Hypogammaglobulinämie und rezidivierende bakterielle Infektionen in Patienten in der Plateauphase des multiplen Myeloms, die nicht auf eine Pneumokokkenimmunisierung angesprochen haben; Kongenitales AIDS mit rezidivierenden Infektionen:
Die empfohlene Dosis beträgt 0,2 - 0,4 g/kg KG alle 3 - 4 Wochen.
Hypogammaglobulinämie in Patienten nach allogener hämatopoetischer Stammzelltransplantation
Die empfohlene Dosis beträgt 0,2 - 0,4 g/kg alle 3 -_4 Wochen. Die Talspiegel sollten über 5g/L gehalten werden.
Primäre Immunthrombozytopenie:
Es gibt zwei unterschiedliche Behandlungsstrategien:
• 0,8 - 1,0 g/kg KG am 1. Tag, . Diese Dosis kann einmal innerhalb von 3 Tagen wiederholt werden,
• jeweils 0,4 g/kg KG für 2 - 5 Tage.
Die Behandlung kann bei einem Rückfall wiederholt werden.
Guillain Barre Syndrom 0,4 g/kg/Tag für 5 Tage.
Chronisch inflammatorisch demyelinisierende Polyneuropathie:
Initialdosis: 2g/kg verteilt auf 4 aufeinanderfolgenden Tagen. Es wird empfohlen die Initialdosis alle drei bis vier Wochen erneut zu geben, bis ein maximales Ansprechen erreicht wurde. Erhaltungsdosis: diese sollte vom Arzt ermittelt werden. Es wird empfohlen nach Erreichen des maximalen Ansprechens die Dosis zu reduzieren und die Verabreichungsfrequenz anzupassen bis die geringst mögliche, wirksame Erhaltungsdosis definiert wurde.
Es konnte gezeigt werden, dass die Initialdosis in bis zu 7 aufeinanderfolgenden Behandlungen innerhalb von 6 Monaten gut vertragen wird.
Kawasaki Syndrom
1,6 - 2,0 g/kg KG sollten auf mehrere Dosen verteilt über 2 - 5 Tage gegeben werden; oder 2 g/kg KG als Einzeldosis. Die Patienten sollten gleichzeitig mit Acetylsalicylsäure (ASS) behandelt werden.
Die Dosierungsempfehlungen werden in folgender Tabelle zusammengefasst:
|
Indikation |
Dosis |
Häufigkeit der Verabreichung |
|
Substitutionsbehandlung bei primären |
- Initialdosis: | |
|
Immundefekten |
0,4 - 0,8 g/kg - anschließend: 0,2 - 0,8 g/kg |
alle 3 - 4 Wochen, um den IgG-Talspiegel auf mindestens 5 - 6 g/l zu halten |
|
Substitutionsbehandlung bei sekundären Immundefekten |
0,2 - 0,4 g/kg |
alle 3 - 4 Wochen, um den IgG-Talspiegel auf mindestens 5-6 g/L zu halten |
|
Kongenitales AIDS |
0,2 - 0,4 g/kg |
alle 3 - 4 Wochen |
|
Hypogammaglobulinämie (<4g/l) in Patienten nach allogener |
0,2 - 0,4 g/kg |
alle 3 - 4 Wochen um einen IgG |
|
hämatopoetischer Stammzelltransplantation |
Talspiegel von über 5g/L zu erreichen | |
|
Immunmodulation: | ||
|
Primäre Immunthrombozytopenie |
0,8 - 1,0 g/kg |
Tag 1, ggf. gefolgt von der gleichen Dosis innerhalb von 3 Tagen |
|
oder | ||
|
0,4 g/kg/Tag |
über 2 - 5 Tage | |
|
Guillain Barre Syndrom |
0,4 g/kg/Tag |
über 5 Tage |
|
Chronisch inflammatorisch |
Initialdosis |
über 4 aufeinanderfolgende Tage verteilt |
|
demyelinisierende Polyneuropathie (CIDP)* |
2g/kg |
alle 3 - 4 Wochen |
|
Erhaltungsdosis |
Diese muss an die Patientenbedürfnisse angepasst werden, siehe auch weiter oben |
|
Kawasaki Syndrom |
1,6 - 2 g/kg oder |
auf mehrere Dosen verteilt über 2 - 5 Tage, zusammen mit Acetylsalicylsäure |
|
2,0 g/kg |
als Einzeldosis zusammen mit Acetylsalicylsäure |
*Die Dosis beruht auf der angewendeten Dosis in der klinischen Studie mit Ig Vena (siehe Abschnitt 5.1)
Spezielle Patientengruppen:
Die Erfahrung in Patienten, welche älter als 65 Jahre sind, ist begrenzt.
Kinder und Jugendliche
Art und Dauer der Anwendung bei Kinder und Jugendlichen (0-18 Jahre) unterscheidet sich nicht von jenen bei Erwachsenen, da die Dosierung für jede Indikation je nach Körpergewicht erfolgt und dem klinischen Befund der oben genannten Erkrankungen angepasst wird.
CIDP:
Wegen der Seltenheit der Krankheit und der dadurch insgesamt geringen Anzahl an Patienten sind nur limitierte Erfahrungen über die Verwendung von intravenösen Immunglobulinen in Kindern mit CIDP vorhanden. Daher sind nur Daten aus der Literatur verfügbar. Allerdings zeigen alle publizierten Daten einheitlich, dass IVIg Behandlungen gleichermaßen in Erwachsenen und Kindern wirksam sind, so wie es auch für andere bereits etablierte IVIg Indikationen der Fall ist.
Art der Anwendung
Für die intravenöse Anwendung.
Normales Immunglobulin vom Menschen sollte intravenös verabreicht werden mit einer initialen Infusionsgeschwindigkeit von 0,46 - 0,92 ml/kg/Stunde (10 - 20 Tropfen pro Minute) während der ersten 20 - 30 Minuten. Bei guter Verträglichkeit (siehe Abschnitt 4.4) kann die Infusionsgeschwindigkeit stufenweise bis auf ein Maximum von 1,85 ml/kg/Stunde (40 Tropfen/Minute) erhöht werden. Siehe auch Abschnitt 6.6.
4.3 Gegenanzeigen
Überempfindlichkeit gegen den Wirkstoff oder einen der sonstigen Bestandteile, welche in Abschnitt 6.1 aufgezählt werden (siehe Abschnitt 4.4).
Überempfindlichkeit gegen Immunglobuline vom Menschen, insbesondere in Patienten, welche Antikörper gegen IgA aufweisen.
4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Dieses Arzneimittel beinhaltet 100 mg Maltose pro Milliliter als sonstigen Bestandteil. Maltose kann manche Blutzuckermesstests beeinflussen und zu fälschlich erhöhten Glucosewerten und damit zu einer unangemessenen Insulinverabreichung führen. Dies wiederum kann eine
lebensbedrohlichen Hypoglykämie (Unterzuckerung) und gegebenenfalls den Tod auslösen. Zudem können Fälle einer echten Hypoglykämie unbemerkt bleiben, wenn der Grad der Unterzuckerung durch fälschlich erhöhte Blutzuckerwerte verschleiert bleibt. Für weitere Informationen siehe Abschnitt 4.5. Hinsichtlich akuten Nierenversagens siehe weiter unten.
Gewisse schwere Unverträglichkeitsreaktionen können mit der Infusionsgeschwindigkeit zusammenhängen. Die unter Punkt 4.2 empfohlene Infusionsgeschwindigkeit sollte daher genau befolgt werden. Die Patienten sind während der gesamten Infusionsdauer genau zu überwachen und in Hinblick auf eventuell auftretende Symptome zu beobachten.
Bestimmte Nebenwirkungen können häufiger auftreten bei:
• hoher Infusionsgeschwindigkeit
• Patienten, die normales Immunglobulin vom Menschen das erste Mal erhalten, oder in seltenen Fälle, bei einem Arzneimittelwechsel, oder wenn die Behandlung für längere Zeit unterbrochen wurde.
Mögliche Komplikationen können oft vermieden werden, wenn:
• durch sehr langsame Erstinfusion (0,46 - 0,92 ml/kg KG/Stunde) sichergestellt wird, dass der Patient nicht gegen normales Immunglobulin vom Menschen sensibilisiert ist;
• sichergestellt ist, dass der Patient über die Infusionsdauer sorgfältig auf etwaige Symptome überwacht wird. Insbesondere sollten Patienten, die das erste Mal Immunglobulin vom Menschen erhalten, bei Arzneimittelwechsel oder nach einer längeren Therapieunterbrechung, sorgfältig für die Dauer der Erstinfusion und während der ersten Stunde nach der Erstinfusion überwacht werden, um mögliche Nebenwirkungen zu bemerken. Alle anderen Patienten sollten nach der Verabreichung mindestens 20 Minuten unter Beobachtung bleiben.
Bei unerwünschter Reaktion muss entweder die Infusionsgeschwindigkeit reduziert werden oder die Infusion abgebrochen werden. Die erforderliche Behandlung richtet sich nach Art und Schwere der unerwünschten Reaktion.
Bei einem Schock sind die aktuellen medizinischen Standardmaßnahmen für eine Schockbehandlung anzuwenden.
Bei allen Patienten erfordert die Gabe von IVIg:
• adäquate Hydratation vor Beginn der IVIg-Infusion
• Überwachung der Urinausscheidung
• Überwachung des Serumkreatinin-Spiegels
• Vermeidung der gleichzeitigen Gabe von Schleifendiuretika.
Überempfindlichkeit
Echte Überempfindlichkeitsreaktionen sind selten. Sie können in Personen mit Anti-IgA-Antikörpern auftreten.
IVIg wird in Patienten mit selektivem IgA Mangel nicht empfohlen, wenn der IgA Mangel die einzige besorgniserregende Krankheit darstellt.
Selten kann normales Immunglobulin vom Menschen eine anaphylaktische Reaktion mit Blutdruckabfall hervorrufen, dies sogar bei Patienten, die die Behandlung bisher gut vertragen haben.
Thromboembolie
Es gibt klinische Hinweise auf einen Zusammenhang zwischen iv. Immunglobulin (IVIg) Gaben und thromboembolischen Ereignissen wie Herzinfarkt, Hirndurchblutungsstörungen (inklusive Schlaganfall), Lungenembolie und tiefer Venenthrombose, dies ist möglicherweise auf eine entsprechende Erhöhung der Blutviskosität durch Immunglobuline bei Risikopatienten zurückzuführen. Die Verordnung und Anwendung von IVIg sollte vorsichtig abgewogen werden bei adipösen Patienten und solchen mit vorbestehenden Risikofaktoren für thrombotische Ereignisse (wie fortgeschrittenes Alter, Bluthochdruck, Diabetes mellitus und Gefäßkrankheiten oder Thrombosen in der Anamnese; ebenso bei Patienten mit erworbener oder angeborener Thrombophilie; bei Patienten mit lang andauernder Immobilisation; mit schwerer Hypovolämie; mit Krankheiten, die zu einer Erhöhung der Blutviskosität führen).
In Patienten mit erhöhtem Risiko für thromboembolische Ereignisse sollten Infusionen von IVIg Produkten mit der geringsten Infusionsrate und in der geringsten noch sinnvollen Dosis erfolgen.
Akutes Nierenversagen
Es gibt Berichte über Fälle von akutem Nierenversagen bei Patienten, die IVIg erhalten haben. In fast allen Fällen konnten Risikofaktoren identifiziert werden, wie vorbestehende Niereninsuffizienz, Diabetes mellitus, Hypovolämie, Übergewicht, gleichzeitige Gabe nephrotoxischer Medikamente oder ein Lebensalter über 65 Jahre.
Bei Verschlechterung der Nierenfunktion sollte man den Abbruch der IVIg Behandlung in Erwägung ziehen. Berichte von eingeschränkter Nierenfunktion und akutem Nierenversagen liegen für viele zugelassene Immunglobuline vor, welche unterschiedliche sonstige Bestandteile wie Saccharose, Glucose und Maltose beinhalten. Jene Präparate, die Saccharose als Stabilisator enthalten, sind jedoch in einem unverhältnismäßig hohen Anteil dafür verantwortlich. Daher sollte bei Risikopatienten die Verwendung von IVIg Produkten, welche keine derartigen sonstigen Bestandteile enthalten überlegt werden. Ig Vena enthält Maltose (siehe sonstige Bestandteile weiter oben).
Zusätzlich sollte bei Patienten mit bekanntem Risiko für ein akutes Nierenversagen das Präparat mit der geringstmöglichen Dosierung und Infusionsrate verabreicht werden.
Aseptisches Meningitis-Syndrom (AMS)
Es gibt Berichte über Fälle von aseptischem Meningitis-Syndrom in Patienten, die IVIg erhalten haben. Das Beenden der IVIg Behandlung brachte eine Besserung des AMS innerhalb weniger Tage ohne weitere Folgen. Die Erkrankung beginnt normalerweise innerhalb weniger Stunden bis 2 Tage nach der IVIg Therapie. Liquoruntersuchungen sind oft positiv für Pleozytose mit bis zu mehreren tausend Zellen pro mm3, vorallem der Granulozytenfamilie, und erhöhtem Proteingehalt mit bis zu mehreren hundert mg/dl.
AMS kann häufiger im Zusammenhang mit Hoch-Dosis (2g/kg KG) IVIg Behandlungen auftreten. Hämolytische Anämie
IVIg-Produkte können Blutgruppen-Antikörper enthalten, welche als Hämolysine wirken und daher eine in vivo Anlagerung von Immunglobulinen an Erthrozyten hervorrufen können. Dies kann zu einem positiven Antiglobulin-Test (Coombs-Test) und, in seltenen Fällen, zur Hämolyse führen. Durch den vermehrten Abbau von roten Blutzellen kann es im Anschluß an eine IVIg Therapie zu einer hämolytischen Anämie kommen. IVIg Empfänger sollten daher genau auf klinische Anzeichen und Symptome einer Hämolyse beobachtet werden (siehe auch Abschnitt 4.8).
Auswirkungen auf serologische Untersuchungen
Nach Verabreichung von Immunglobulinen kann es durch den vorübergehenden Anstieg der verschiedenen passiv übertragenen Antikörper im Blut des Patienten zu falschen Testergebnissen bei serologischen Untersuchungen kommen.
Die passive Übertragung von Antikörpern gegen Erythrozytenantigene, z. B. A, B, D, kann einige serologische Tests auf Erythrozyten-Antikörper wie zum Beispiel den direkten Antiglobulin-Test (DAT, direkter Coombs-Test) beeinträchtigen.
Übertragbare Erreger
Zu den Standardmaßnahmen, um Infektionen aufgrund der Verwendung von Arzneimitteln aus menschlichem Blut oder Plasma zu verhindern, zählen: Spenderauswahl, Testung einzelner Spenden und des Plasmapools auf bestimmte Infektionsmarker und schließen effektive Virusinaktivierungs-/Viruseliminierungsverfahren im Herstellungsprozess ein.
Trotz dieser Maßnahmen kann bei der Verabreichung von Arzneimitteln aus menschlichem Blut oder Blutplasma die Übertragung von Krankheitserregern nicht völlig ausgeschlossen werden. Dies gilt auch für unbekannte oder neu auftretende Viren oder andere Pathogene.
Die verwendeten Verfahren werden bei umhüllten Viren wie HIV, HBV und HCV und dem nicht umhüllten Virus HAV als ausreichend erachtet.
Bei nicht umhüllten Viren wie Parvovirus B19 sind sie möglicherweise von eingeschränktem Wert.
Die klinischen Erfahrungen zeigen ein geringes Risiko hinsichtlich einer Übertragung von Hepatitis A oder Parvovirus B19 durch Immunglobuline; man geht davon aus, dass der Antikörpergehalt der Produkte einen wesentlichen Beitrag zur Virensicherheit leistet.
Es wird dringend empfohlen, jede Verabreichung von Ig Vena mit Arzneimittelnamen und Chargennummer zu dokumentieren, um die Verbindung zwischen Patient und Charge herstellen zu können.
Kinder und Jugendliche
Es sind keine zusätzlichen spezifischen Maßnahmen oder spezifische Überwachung von Kindern und Jugendlichen notwendig.
4.5 Wechselwirkung mit anderen Arzneimitteln und sonstige Wechselwirkungen
Lebendvirusimpfstoffe
Die Verabreichung von Immunglobulinen kann die Wirkung von Virus-Lebendimpfstoffen wie Masern, Röteln, Mumps und Varizellen über einen Zeitraum von mindestens 6 Wochen bis zu 3 Monaten beeinträchtigen. Nach Verabreichung des Arzneimittels soll daher ein Zeitraum von 3 Monaten verstreichen, bevor eine Impfung mit einem Lebendvirusimpfstoff erfolgt. Bei Masern kann dieser Zeitraum bis zu einem Jahr andauern. Deshalb sollten bei Patienten, die eine Masernimpfung erhalten haben, die Antikörperspiegel überprüft werden.
Blutzuckermesstests
Bestimmte Blutzuckermesstests (zum Beispiel jene, welche auf Glucose-Dehydrogenase-Pyrrolochinolinchinon (GDH-PQQ) oder Glucose-Dye-Oxidoreduktase basieren) erkennen fälschlicherweise die in Ig Vena enthaltene Maltose (100mg/ml) als Glucose. Das kann zu fälschlich erhöhten Glucosemesswerten während und bis zu 15 Stunden nach einer Infusion führen und somit zu einer unangemessenen Insulinverabreichung, welche wiederum in einer lebensbedrohlichen oder sogar fatalen Hypoglykämie enden kann.
Zudem können Fälle einer echten Hypoglykämie unbemerkt bleiben, wenn der Grad der Unterzuckerung durch fälschlich erhöhte Blutzuckerwerte verschleiert wird. Demzufolge muss bei Verabreichung von Ig Vena oder anderen parenteralen maltosehältigen Produkten, der Blutzuckergehalt mittels einer glucosespezifischen Testmethode bestimmt werden.
Die Gebrauchsinformation des Blutzuckertests sowie jene der Teststreifen sollte sorgfältig gelesen werden, um festzustellen, ob der Test für die gleichzeitige Verabreichung von maltosehältigen, parenteralen Produkten geeignet ist. Bei Unklarheit, wenden Sie sich an den Hersteller der Testmethode, um zu erfahren, ob der Test bei gleichzeitiger Verabreichung von maltosehältigen, parenteralen Produkten geeignet ist.
Kinder und Jugendliche
Obwohl keine spezifischen Wechselwirkungsstudien bei Kindern und Jugendlichen durchgeführt wurden, sind keine Unterschiede zur Erwachsenenpopulation zu erwarten.
4.6 Fertilität, Schwangerschaft und Stillzeit
Schwangerschaft
Es liegen keine kontrollierten klinischen Studien zur Unbedenklichkeit der Anwendung in der Schwangerschaft vor. Die Verabreichung des Arzneimittels an schwangere Frauen oder stillende Mütter sollte deshalb sorgfältig abgewogen werden. Es wurde gezeigt, dass IVIg Produkte die Plazenta durchwandern können, vor allem im dritten Trimester.
Die lange klinische Erfahrung mit Immunglobulinen lässt erkennen, dass keine schädigende Wirkung auf den Verlauf der Schwangerschaft, den Fötus oder das Neugeborene zu erwarten ist.
Stillzeit
Immunglobuline gehen in die Muttermilch über und können dazu beitragen, das Neugeborene vor jenen Pathogenen zu schützen, welche über die Schleimhäute eindringen.
Fertilität
Klinische Erfahrungen mit Immunglobulinen deuten darauf hin, dass keine schädlichen Einflüsse auf die Fertilität erwartet werden müssen.
4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Manche unerwünschte Wirkungen von Ig Vena können die Fahrtüchtigkeit und die Fähigkeit zum Bedienen von Maschinen beeinflussen. Patienten, bei denen unerwünschte Arzneimittelreaktionen auftreten, sollten abwarten bis diese vollständig überwunden sind, bevor sie selbst fahren oder Maschinen bedienen.
4.8 Nebenwirkungen
Zusammenfassung des Sicherheitsprofils
Manchmal können Schüttelfrost, Kopfschmerz, Schwindel, Fieber, Erbrechen, allergische Reaktionen, Übelkeit, Gelenkschmerzen, Blutdruckabfall und leichte Rückenschmerzen vorkommen.
Selten können normale Immunglobuline vom Menschen einen plötzlichen Blutdruckabfall verursachen und in Einzelfällen kann ein anaphylaktischer Schock auftreten, auch wenn der Patient bei früheren Anwendungen keine Reaktion gezeigt hat.
Fälle reversibler aseptischer Meningitis, und seltene Fälle vorübergehender kutaner Reaktionen wurden bei der Gabe von normalem Immunglobulin vom Menschen beobachtet. In manchen Patienten wurden Fälle von reversiblen hämolytischen Reaktionen beobachtet, besonders in jenen, welche die Blutgruppe A, B und AB hatten. Selten können nach Hoch-Dosis- IVIG Behandlungen hämolytische Reaktionen auftreten, welche eine Transfusion erforderlich machen.
Ein Anstieg von Serumkreatinin und/oder ein akutes Nierenversagen wurden beobachtet.
Sehr selten sind thromboembolische Zwischenfälle wie Herzinfarkt, Schlaganfall, Lungenembolie und tiefe Venenthrombose beobachtet worden.
Tabellarische Zusammenfassung der unerwünschten Reaktionen:
Die folgende Tabelle bezieht sich auf die Systemorganklassen gemäß MedDRA Datenbank (SOC und bevorzugter Begriff). Die Häufigkeiten wurden gemäß folgender Einteilung bestimmt: sehr häufig (> 1/10), häufig (>1/100 bis <1/10), gelegentlich (>1/1.000 bis <1/100); selten (>1/10.000 bis <1/1.000); sehr selten (<1/10.000), nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
Es gibt keine soliden Daten aus klinischen Studien über die Häufigkeit von Nebenwirkungen. Die folgenden Informationen basieren auf Berichten aus der Post-Marketing Überwachung:
|
MedDRA Systemorganklasse |
Bevorzugter Begriff | |
|
(SOC) |
gemäß MedDRA |
Häufigkeit |
|
Erkrankungen der Atemwege, des Brustraumes und des Mediastinums |
Lungenembolie |
nicht bekannt |
|
Erkrankungen des Nervensystems |
Schlaganfall |
nicht bekannt |
|
Kopfschmerz |
sehr selten | |
|
Schwindel |
sehr selten | |
|
Herzerkrankungen |
Myokardinfarkt |
nicht bekannt |
|
Gefäßerkrankungen |
Tiefe Venenthrombose |
nicht bekannt |
|
Embolie |
nicht bekannt | |
|
Hypotonie |
sehr selten | |
|
Erkrankungen des Immunsystems |
Anaphylaktischer Schock |
nicht bekannt |
|
Hypersensibilität |
nicht bekannt | |
|
Erkrankungen der Nieren- und Harnwege |
Akutes Nierenversagen |
sehr selten |
|
Infektionen und parasitäre Erkrankungen |
aseptische Meningitis |
sehr selten |
|
Erkrankungen des Blutes und des Lymphsystems |
Hämolyse |
sehr selten |
|
Hämolytische Anämie |
nicht bekannt | |
|
Erkrankungen der Haut und des Unterhautzellgewebes |
Hautreaktionen |
sehr selten |
|
Skelettmuskulatur-,Bindegewebs- und Knochenerkrankungen |
Arthralgie |
sehr selten |
|
Rückenschmerzen |
sehr selten | |
|
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort |
Fieber |
sehr selten |
|
Schüttelfrost |
sehr selten | |
|
Erkrankungen des Gastrointestinaltraktes |
Erbrechen |
sehr selten |
|
Übelkeit |
sehr selten | |
|
Untersuchungen |
Blutdruck erniedrigt |
nicht bekannt |
|
Kreatinin im Blut erhöht |
nicht bekannt |
Häufigkeit der Unerwünschten Arzneimittelwirkungen (UAW) während der klinischen CIDP Studie mit Ig Vena (Ig Vena wurde in dieser Studie 756 mal verabreicht)
|
MedDRA Systemorganklasse (SOC) |
Bevorzugter Begriff gemäß MedDRA |
Häufigkeit |
|
Erkrankungen des Nervensystems |
Kopfschmerz |
Gelegentlich (1 in 756 Verabreichungen) |
Informationen zur Sicherheit hinsichtlich übertragbarer Erreger finden sich unter 4.4.
Kinder und Jugendliche
Es wird erwartet, dass sich Häufigkeit, Art und Schweregrad von unerwünschten Wirkungen in Kindern und Jugendlichen nicht von denen in erwachsenen Patienten unterscheiden.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung in Deutschland über das Paul-Ehrlich-Institut, Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel, Pharmakovigilanz II, Paul-Ehrlich-Straße 51-59, D-63225 Langen, Tel: +49 6103 77 0, Fax: +49 6103 77 1234, Website: http://www.pei.de und in Österreich über das Bundesamt für Sicherheit im Gesundheitswesen, Inst. Pharmakovigilanz, Traisengasse 5, 1200 WIEN, Fax: + 43 (0) 50 555 36207, Website: http://www.basg.gv.at anzuzeigen.
4.9 Überdosierung
Überdosierung kann zu Volumenüberlastung und Hyperviskosität führen, besonders bei Risikopatienten einschließlich älteren Patienten, und bei Patienten mit eingeschränkter Herz- oder Nierenfunktion.
Dabei wird nicht erwartet, dass es Unterschiede zwischen Erwachsenen und der pädiatrischen Population (0-18 Jahre) gibt.
5 PHARMAKOLOGISCHE EIGENSCHAFTEN
5.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Immunsera und Immunglobuline: Immunglobuline, normal human, zur intravasalen Anwendung; ATC-Code: J06BA02.
Normales Immunglobulin vom Menschen enthält hauptsächlich Immunglobulin G (IgG) mit einem breiten Spektrum an Antikörpern gegen Infektionserreger.
Normales Immunglobulin vom Menschen enthält das komplette Antikörperspektrum an funktionell intaktem Immunglobulin G, das in der Normalbevölkerung vorhanden ist. Es wird aus gepooltem Plasma von mindestens 1000 Spendern hergestellt. Die Verteilung der Immunglobulin G-Subklassen entspricht nahezu der des natürlichen, menschlichen Plasmas. Durch Verabreichung entsprechender Dosen von Ig Vena werden abnormal verminderte IgG-Spiegel wieder in den Normalbereich angehoben.
Der Wirkmechanismus von Immunglobulinen bei anderen Indikationen als der Substitutionstherapie ist nicht vollständig aufgeklärt, schließt aber immunmodulatorische Effekte mit ein.
Kinder und Jugendliche
Die publizierten Daten aus Studien zu Wirksamkeit und Sicherheit haben keine Unterschiede zwischen Kindern und Erwachsenen, welche an derselben Erkrankung leiden, ergeben.
Klinische Studien mit Ig Vena in Patienten mit chronisch inflammatorisch demyelinisierender Polyneuropathie (CIDP):
Die doppelblinde kontrollierte Phase III Studie (KB034), welche Verträglichkeit und Wirksamkeit der Langzeit-Behandlung mit hohen Dosen an intravenösen Immunglobulinen mit der LangzeitBehandlung mit hohen Dosen an intravenösen Methylprednisolon (IVMP) vergleicht, wurde in insgesamt 46 erwachsenen CIDP-Patienten durchgeführt. Die Patienten wurden randomisiert und erhielten entweder Ig Vena (Dosierung 2g/kg/Monat verteilt auf 4 aufeinanderfolgende Tage, 6 Monate lang) oder IVMP (Dosierung: 2g/Monat verteilt auf 4 aufeinanderfolgende Tage, 6 Monate lang).
10 von 21 (47,6%) mit IVMP behandelten Patienten beendeten die 6-monatige Behandlung im Vergleich zu 21 von 24 (87,5%) mit Ig Vena behandelten Patienten (p=0.0085). Die kumulierte Wahrscheinlichkeit eines Behandlungsabbruches nach 15 Tagen, 2 und 6 Monaten, war signifikant höher bei Behandlung mit IVMP als bei Behandlung mit Ig Vena.
Von den 11 IVMP Patienten, welche ihre Behandlung abbrachen, taten dies 8 wegen zunehmender Verschlechterung nach Behandlungsbeginn (5 Patienten) oder wegen fehlender Verbesserung nach 2 Behandlungszyklen (3 Patienten). 1 Patient brach die Behandlung wegen Nebenwirkungen (Gastritis) ab (9,1%) und 2 zogen ihre Zustimmung freiwillig zurück (18,2%).
In der IVIG Gruppe beendeten 3 Patienten die Behandlung frühzeitig wegen zunehmender Verschlechterung: 2 nach Therapiebeginn und 1 Patient wegen fehlender Verbesserung nach 2 Behandlungszyklen.
Alle Patienten, welche wegen zunehmender Verschlechterung oder fehlender Verbesserung die Behandlung abbrachen, wurden mit der Alternativtherapie weiterbehandelt. Die drei Patienten aus der IVMP Gruppe, die die Behandlung wegen Nebenwirkungen oder Zurücknahme der Einwilligung abbrachen, verweigerten eine weitere Behandlung.
Die Ergebnisse zu den sekundären Studienendpunkten werden in der nachfolgenden Tabelle zusammengefasst (statistisch signifikante Unterschiede sind fett gedruckt):
|
Intention To Treat Population (ITT) |
Per Protocol Population (PP) | |||||
|
Secondary endpoints |
Ig Vena® 10 g/200 ml |
IVMP |
p- value |
Ig Vena® 10 g/200 ml |
IVMP |
p- value |
|
Relapses rate * |
45.8% (n 11/24) |
52.4% (n 11/21) |
0.7683 |
38.1% (n 8/21) |
0% (n 0/10) |
0.0317 |
|
MRC sum score [delta (p-value)] |
+4.7 (0.0078) |
+1.8 (0.1250) |
0.6148 |
+4.0 (0.0469) |
+2.0 (0.5000) |
0.5473 |
|
INCAT (p-value) |
0.0004 |
0.1877 |
0.3444 |
0.0057 |
0.2622 |
0.9065 |
|
Vibratory score - Right medial malleolus (p-value) |
<0.0001 |
0.6515 |
0.0380 |
0.0009 |
0.2160 |
0.4051 |
|
Fist strength right [delta (p-value)] |
+19.4 (0.0005) |
+5.4 (0.6169) |
0.0641 |
+16.5 (0.0044) |
+14.7 (0.0156) |
0.5012 |
|
Fist strength left [delta (p-value)] |
+16.9 (0.0011) |
+8.8 (0.1170) |
0.1358 |
+12.7 (0.0014) |
+10.5 (0.0156) |
0.3330 |
|
Time on 10 meters [delta (p-value)] |
-3.2 (0.0025) |
-0.5 (0.2051) |
0.0800 |
-3.5 (0.0043) |
-2.0 (0.4453) |
0.2899 |
|
ONLS scale (p-value) |
0.0006 |
0.0876 |
0.4030 |
0.0033 |
0.0661 |
0.8884 |
|
Rankin scale (p-value) |
0.0006 |
0.0220 |
0.3542 |
0.0132 |
0.2543 |
0.8360 |
|
Rotterdam scale [delta (p-value)] |
+1.4 (0.0071) |
+1.3 (0.0342) |
0.6465 |
+1.1 (0.0342) |
+1.1 (0.0859) |
0.4056 |
|
SF-36 QoL |
+14.2 (0.0011) |
+16.7 (0.0008) |
0.3634 |
+11.1 (0.0091) |
+16.0 (0.1094) |
0.6518 |
*ITT: während der gesamten Studie (12 Monate); PP: Follow-Up Phase (6 Monate)
5.2 Pharmakokinetische Eigenschaften
Normales Immunglobulin vom Menschen ist nach intravenöser Applikation sofort und vollständig im Kreislauf des Patienten bioverfügbar. Es verteilt sich relativ schnell zwischen Plasma und extravaskulärer Flüssigkeit; das Gleichgewicht zwischen Intra- und Extravasalraum ist nach etwa 3 - 5 Tagen erreicht.
Normales Immunglobulin vom Menschen hat eine Halbwertszeit von 21 Tagen. Diese kann von Patient zu Patient variieren, besonders bei primären Immundefekten.
IgG und IgG-Komplexe werden in den Zellen des retikuloendothelialen Systems abgebaut.
Kinder und Jugendliche
Die publizierten Daten aus pharmakokinetischen Studien haben keinen Unterschied zwischen Kindern und Erwachsenen, welche an derselben Erkrankung leiden, ergeben. Es liegen keine Daten aus pharmakokinetischen Studien über pädriatische Patienten mit CIDP vor.
5.3 Präklinische Daten zur Sicherheit
Immunglobuline sind normale Bestandteile des menschlichen Körpers. Präklinische Daten zur Sicherheit aus dem Tiermodell sind auf Grund der Induktion und Interferenz von Antikörpern gegen heterologe Proteine limitiert. Trotz ihres eingeschränkten Nutzens lassen Tierstudien zur akuten und subakuten Toxizität keine besonderen Gefahren für den Menschen erkennen.
6 PHARMAZEUTISCHE ANGABEN
6.1 Liste der sonstigen Bestandteile
Maltose
Wasser für Injektionszwecke
6.2 Inkompatibilitäten
Da keine Kompatibilitätsstudien durchgeführt wurden, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
6.3 Dauer der Haltbarkeit
3 Jahre für die Packungsgröße 100 ml.
2 Jahre für die Packungsgrößen 20, 50 und 200 ml.
Geöffnete Infusionsflaschen sind umgehend zu verbrauchen.
6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Im Kühlschrank lagern (2°C - 8°C), Durchstechflasche im Umkarton aufbewahren.
Vor Gebrauch und innerhalb der Haltbarkeitsdauer können die Durchstechflaschen mit 50, 100 und 200 ml für maximal 6 aufeinander folgende Monate bei Raumtemperatur, maximal 25°C, gelagert werden. Nach dieser Zeitspanne muss das Produkt entsorgt werden. In keinem Fall darf das Produkt wieder in den Kühlschrank gestellt werden, wenn es einmal bei Raumtemperatur gelagert worden ist.Der Beginn der Raumtemperaturlagerung muss auf dem äußeren Karton vermerkt werden.
Nicht einfrieren.
6.5 Art und Inhalt des Behältnisses
20 ml Lösung in einer Durchstechflasche (Type I Glas) mit Gummistopfen (Halobutyl-Gummi); Packungsinhalt: eine Durchstechflasche.
50 ml, 100 ml und 200 ml Lösung in einer Durchstechflasche (Type I Glas) mit Gummistopfen (Halobutyl- Gummi); Packungsinhalt: eine Durchstechflasche + Aufhängevorrichtung.
6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung
Vor der Verabreichung sollte das Produkt auf Körper- oder Raumtemperatur angewärmt werden.
Die Lösung sollte klar oder leicht opaleszierend, farblos oder leicht gelblich sein.
Lösungen, die trüb sind oder Ablagerungen aufweisen, dürfen nicht verwendet werden.
Vor Verabreichung soll eine Sichtprüfung der Infusionslösung auf ungelöste Partikel und Verfärbungen stattfinden.
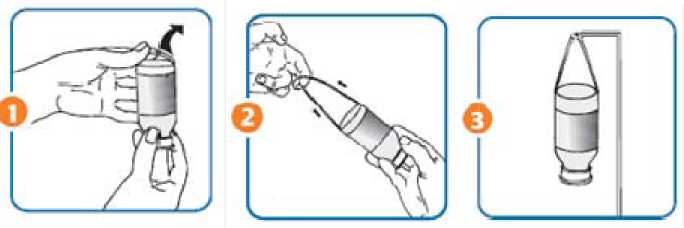
Anleitung zur Verwendung der Aufhängevorrichtung
1. Den integrierten Aufhängebügel am unteren Rand des Etikettes lösen und nach oben ziehen. (Abb.A)
2. Falls notwendig, den Aufhängebügel durch Ziehen verlängern. (Abb.B)
3. Die Flasche mit dem Aufhängebügel am Infusionsständer anbringen. (Abb.C)
( Abb. A) (Abb. B) (Abb. C)
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu entsorgen.
INHABER DER ZULASSUNG
7
Kedrion S.p.A.
Loc. Ai Conti
55051 Castelvecchio Pascoli Barga (Lucca)
Italien.
8 ZULASSUNGSNUMMER
AT: Z.Nr.: 2-00321
DE: PEI.H.03409.01.1
Ig Vena 50 g/l Infusionslösung Ig Vena 50 g/l Infusionslösung Ig Vena 50 g/l Infusionslösung Ig Vena 50 g/l Infusionslösung
20 ml Flasche
50 ml Flasche + Aufhängevorrichtung 100 ml Flasche + Aufhängevorrichtung 200 ml Flasche + Aufhängevorrichtung
9 DATUM DER ERTEILUNG DER ZULASSUNG/VERLANGERUNG DER ZULASSUNG
AT: 22.02.2007/ 01.06.2010
DE: 02.02.2007/08.10.2010
10 STAND DER INFORMATION
06/11/2014
DE: HERKUNFTSLÄNDER DES BLUTPLASMAS
Deutschland, Österreich, USA, Polen, Ungarn und Tschechien.
VERSCHREIBUNGSPFLICHT / APOTHEKENPFLICHT
AT: Rezept- und apothekenpflichtig, wiederholte Abgabe verboten. DE: Verschreibungspflichtig
16