Immunate Stim Plus 250 I.e. Immuno
alt informationenGebrauchsinformation: Information für Anwender
IMMUNATE STIM plus 250 I.E./500 I.E./1000 I.E. Immuno
Pulver und Lösungsmittel zur Herstellung einer Injektionslösung
Lesen Sie die gesamte Packungsbeilage sorgfältig durch, bevor Sie mit der Anwendung dieses Arzneimittels beginnen, denn sie enthält wichtige Informationen.
• Heben Sie die Packungsbeilage auf. Vielleicht möchten Sie diese später nochmals lesen.
• Wenn Sie weitere Fragen haben, wenden Sie sich an Ihren Arzt oder Apotheker.
• Dieses Arzneimittel wurde Ihnen persönlich verschrieben. Geben Sie es nicht an Dritte weiter.
Es kann anderen Menschen schaden, auch wenn diese die gleichen Beschwerden haben wie Sie.
• Wenn Sie Nebenwirkungen bemerken, wenden Sie sich an Ihren Arzt oder Apotheker. Dies gilt auch für Nebenwirkungen, die nicht in dieser Packungsbeilage angegeben sind. Siehe Abschnitt 4.
Was in dieser Packungsbeilage steht
1. Was ist IMMUNATE und wofür wird es angewendet?
2. Was sollten Sie vor der Anwendung von IMMUNATE beachten?
3. Wie ist IMMUNATE anzuwenden?
4. Welche Nebenwirkungen sind möglich?
5. Wie ist IMMUNATE aufzubewahren?
6. Inhalt der Packung und weitere Informationen
1. Was ist IMMUNATE und wofür wird es angewendet?
Dieses Arzneimittel ist ein Kombinationspräparat aus Blutgerinnungsfaktor VIII vom Menschen und von-Willebrand-Faktor (vWF:RCoF) vom Menschen zur intravenösen Anwendung.
IMMUNATE wird angewendet zur
• Behandlung und Prophylaxe von Blutungen bei Patienten mit angeborenem oder erworbenem Faktor VIII-Mangel (Hämophilie A, Hämophilie A mit Faktor VIII-Inhibitor, erworbener Faktor VIII-Mangel aufgrund einer spontanen Entwicklung von Faktor VIII-Inhibitor).
• Behandlung von Blutungen bei Patienten mit von-Willebrand-Syndrom mit Blutgerinnungsfaktor VIII-Mangel, wenn kein spezifisches bei von-Willebrand-Syndrom wirksames Plasmapräparat zur Verfügung steht.
2. Was sollten Sie vor der Anwendung von IMMUNATE beachten?
IMMUNATE darf nicht angewendet werden,
• bei Überempfindlichkeit gegenüber einem der arzneilich wirksamen Bestandteile oder einem der sonstigen Bestandteile.
Warnhinweise und Vorsichtsmaßnahmen
Wenn während der Verabreichung von IMMUNATE Überempfindlichkeitsreaktionen auftreten, ist die Injektion/Infusion sofort abzubrechen und eine entsprechende medizinische Behandlung einzuleiten. Frühe Anzeichen von Überempfindlichkeitsreaktionen sind z. B. Nesselsucht, generalisierte Urtikaria, beschleunigter Herzschlag, Schmerzen in der Brust, Atembeschwerden, Ödeme (inklusive Gesichts- und Lidödeme), Hautausschlag, Gesichtsrötung, Juckreiz, niedriger Blutdruck sowie Anaphylaxie bis hin zum anaphylaktischen Schock. Die aktuellen medizinischen Richtlinien zur Schocktherapie sind zu beachten.
Bei der Herstellung von Arzneimitteln aus Blut oder Blutplasma werden bestimmte Maßnahmen getroffen, um der Übertragung von Infektionen auf die Patienten vorzubeugen. Diese schließen eine sorgfältige Auswahl der Blut- und Plasmaspender ein, um sicherzustellen, dass Risikospender, die Träger von Infektionserregern sein können, ausgeschlossen werden. Weiterhin schließt dies die Testung der einzelnen Blutspenden und Plasmapools auf Virus-/und Infektionsmarker ein. Die Hersteller dieser Produkte schließen auch Verfahrensschritte bei der Produktion von Arzneimitteln aus Blut und Blutplasma mit ein, die Viren inaktivieren oder eliminieren können. Trotz dieser Maßnahmen kann die Möglichkeit der Infektionsübertragung bei der Verabreichung von Arzneimitteln aus Blut oder Blutplasma nicht völlig ausgeschlossen werden. Dies trifft auch für bislang unbekannte oder neu aufgetretene Viren oder andere Arten von Infektionen zu.
Die Maßnahmen werden als effektiv gegen umhüllte Viren wie das AIDS-Virus (HIV), gegen das Hepatitis B-Virus und gegen das Hepatitis C-Virus sowie gegen das nicht-umhüllte Virus wie das Hepatitis A-Virus (HAV) betrachtet. Die Maßnahmen können möglicherweise gegen nicht-umhüllte Viren wie das Parvovirus B19 nur begrenzt wirksam sein. Eine Parvovirus B19-Infektion kann bei Schwangeren schwerwiegende Folgen haben (Infektion des Fötus) und bei Menschen, die an einem Immundefekt oder einem erhöhten Erythrozytenabbau leiden (z. B. Sichelzellanämie oder hämolytische Anämie).
Ihr behandelnder Arzt wird Ihnen möglicherweise die Impfung gegen Hepatitis A und B empfehlen, wenn Sie regelmäßig/oder wiederholt aus Blutplasma hergestellte Faktor VIII-Konzentrate/von-Willebrand-Faktoren-Konzentrate, wie IMMUNATE erhalten.
Es wird dringend empfohlen, dass bei jeder Verabreichung von IMMUNATE Name und Chargenbezeichnung des Präparates dokumentiert werden, um die Aufzeichnung der verwendeten Produktcharge sicherstellen zu können.
Bei der Behandlung von Patienten mit von-Willebrand-Syndrom besteht ein Risiko hinsichtlich des Auftretens thrombotischer Ereignisse, besonders bei Vorliegen von bekannten klinischen oder labortechnisch belegten Risikofaktoren. Deshalb müssen Risikopatienten auf frühe Zeichen von Thrombosen hin überwacht werden. Eine Prophylaxe gegenüber Thromboembolien sollte gemäß den aktuellen Empfehlungen durchgeführt werden. Bei Patienten, die mit einem von-Willebrand-Faktor Produkt behandelt werden, das Faktor VIII enthält, sollte unbedingt der Faktor-VIII:C-Plasmaspiegel überwacht werden, um eine dauerhafte Erhöhung zu vermeiden, da dies mit einem erhöhten Risiko für thrombotische Ereignisse einhergeht.
Bei Patienten, bei denen in der Vergangenheit bereits venöse Thromboembolien aufgetreten sind, wurde ein endogener hoher Faktor-VIII-Spiegel mit einem erhöhten Risiko für spätere thrombotische Ereignisse in Verbindung gebracht.
Patienten mit von-Willebrand-Syndrom, besonders solche mit Typ 3, können neutralisierende Antikörper (Inhibitoren), gegen den von-Willebrand-Faktor entwickeln. Wenn der erwartete Anstieg der von-Willebrand-Ristocetin-Cofaktor-Aktivität im Plasma nicht erreicht wird, oder wenn Blutungen nicht mit einer entsprechenden Dosis beherrscht werden können, sollten geeignete Tests auf die Anwesenheit von von-Willebrand-Inhibitoren durchgeführt werden. Es besteht die Möglichkeit, dass bei Patienten mit hohen Inhibitortitem die von-Willebrand-Therapie nicht effektiv ist und andere therapeutische Möglichkeiten in Betracht gezogen werden sollten. Die Behandlung solcher Patienten sollte unter der Leitung von Ärzten durchgeführt werden, die mit der Behandlung von Patienten mit Gerinnungsstörungen vertraut sind.
Diese Antikörper können auch eine Anaphylaxie auslösen. Patienten, bei denen es zu einer anaphylaktischen Reaktion kommt, sollten daher auf Inhibitoren gegen den von-Willebrand-Faktor untersucht werden.
Die Bildung von Antikörpern mit Faktor VIII-neutralisierender Wirkung (Inhibitoren) ist eine bekannte Komplikation bei der Behandlung von Hämophilie A-Patienten. Diese Inhibitoren sind meist gegen die prokoagulatorische Aktivität von Faktor VIII gerichtete IgG-Immunglobuline, die in Bethesda Einheiten (B.E.) pro ml Plasma (modifizierter Bethesda Assay) quantifiziert werden. Das Risiko einer HemmkörperEntwicklung wird von einer Reihe von Faktoren bestimmt, die mit speziellen Eigenschaften des jeweiligen Patienten zusammenhängen. Als wichtigste Risikofaktoren gelten unter anderem die Art der FVIII-Genmutation, die Familienanamnese und die ethnische Zugehörigkeit.
Über Inhibitoren wurde überwiegend bei nicht vorbehandelten Patienten berichtet.
Das Risiko einer Bildung von Inhibitoren hängt mit der Exposition gegenüber Antihämophilie-Faktor VIII zusammen, wobei dieses Risiko innerhalb der ersten 20 Expositionstage am höchsten ist. In seltenen Fällen können sich Inhibitoren auch nach den ersten 100 Expositionstagen bilden.
Patienten, die mit Blutgerinnungsfaktor VIII vom Menschen behandelt werden, sollten durch entsprechende klinische Überwachungsmethoden und Laboruntersuchungen sorgfältig auf die Bildung von Inhibitoren überwacht werden. Siehe auch Abschnitt 4.
Das Produkt sollte mit Vorsicht bei Kindern unter sechs Jahren angewendet werden, die noch nicht häufig mit Faktor VIII-Produkten behandelt wurden, da systematische klinische Untersuchungen an Kindern nicht durchgeführt wurden.
IMMUNATE enthält Blutgruppen-Isoagglutinine (anti-A und anti-B). Bei Patienten mit den Blutgruppen A, B oder AB kann es nach wiederholter Anwendung in kurzen Abständen oder nach der Anwendung sehr hoher Dosen zu einer Hämolyse kommen. Sehr hohe Dosen innerhalb kurzer Zeit werden möglicherweise im Rahmen einer Immuntoleranztherapie zur Behandlung einer Hämophilie A mit Faktor-VIII-Inhibitor eingesetzt.
Anwendung von IMMUNATE zusammen mit anderen Arzneimitteln
Derzeit sind keine pharmakologischen Wechselwirkungen mit anderen Arzneimitteln bekannt.
Informieren Sie Ihren Arzt oder Apotheker, wenn Sie andere Arzneimittel einnehmen/anwenden, kürzlich andere Arzneimittel eingenommen/angewendet haben oder beabsichtigen, andere Arzneimittel einzunehmen/anzuwenden.
Schwangerschaft und Stillzeit
Wenn Sie schwanger sind oder stillen, oder wenn Sie vermuten, schwanger zu sein oder beabsichtigen, schwanger zu werden, fragen Sie vor Anwendung dieses Arzneimittels Ihren Arzt um Rat.
Reproduktionstoxikologische Studien mit Labortieren wurden nicht durchgeführt. Die Unbedenklichkeit von Blutgerinnungsfaktor VIII vom Menschen bei Schwangerschaften wurde nicht in kontrollierten klinischen Versuchen festgestellt. IMMUNATE ist daher während der Schwangerschaft und Stillzeit nur anzuwenden, wenn dies unbedingt erforderlich ist.
Informationen zu Parvovirus B19-Infektionen siehe unter Abschnitt 2.
Verkehrstüchtigkeit und Fähigkeit zum Bedienen von Maschinen
Es sind keine Auswirkungen von IMMUNATE auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen bekannt.
IMMUNATE enthält Natrium
Bei Patienten, die auf eine natriumarme Ernährung achten müssen, muss der Natriumgehalt berücksichtigt werden, da der Natrium-Gehalt in der maximalen täglichen Dosis eventuell höher ist als 200 mg.
3. Wie ist IMMUNATE anzuwenden?
Dosierung
Wenden Sie dieses Arzneimittel immer genau nach Absprache mit Ihrem Arzt an. Fragen Sie bei Ihrem Arzt oder Apotheker nach, wenn Sie sich nicht sicher sind.
Falls vom Arzt nicht anders verordnet, gelten folgende Angaben:
A) Dosierung bei Hämophilie A:
Dosierung und Dauer der Substitutionstherapie hängen von der Schwere des Faktor VIII-Mangels sowie dem Ort und Ausmaß der Blutung und dem klinischen Zustand des Patienten ab.
Die Anzahl der verabreichten Faktor VIII-Einheiten ist in Internationalen Einheiten (I.E.) angegeben, die sich auf den aktuellen Standard der WHO für Faktor VIII-Produkte beziehen. Die Faktor VIII-Aktivität im Plasma wird entweder prozentual (relativ zu normalem Humanplasma) oder in Internationalen Einheiten (bezogen auf den Internationalen Standard für Faktor VIII-Konzentrate) ausgedrückt.
Eine Internationale Einheit (I.E.) Faktor VIII-Aktivität entspricht der Menge an Faktor VIII in 1 ml normalem Humanplasma.
Die unten angegebene Berechnung der erforderlichen Dosis Faktor VIII beruht auf der empirischen Erkenntnis, dass 1 Internationale Einheit (I.E.) Faktor VIII pro kg Körpergewicht die Faktor VIII-Aktivität im Plasma um ca. 2 % der normalen Aktivität erhöht.
Die erforderliche Dosis wird mit folgender Formel ermittelt:
Erforderliche Einheiten = Körpergewicht (kg) x gewünschter Faktor VIII-Anstieg (%) x 0,5
Die Dosis und das Dosierungsintervall sollten sich immer nach der klinischen Wirksamkeit im Einzelfall richten.
Blutungen und chirurgische Eingriffe
Bei folgenden Blutungsereignissen darf die Faktor VIII-Aktivität im entsprechenden Zeitraum nicht unter die angegebenen Plasmaspiegel sinken.
Die folgende Tabelle enthält Richtwerte für die Dosierung bei Blutungen und chirurgischen Eingriffen:
|
Grad der Blutung/Art des chirurgischen Eingriffs |
Erforderlicher FVIII-Spiegel (% des Normalwerts) (I.E./dl) |
Substitutionsintervall (Stunden) / Behandlungsdauer (Tage) |
|
Blutung | ||
|
Frühstadium von Gelenks- und Muskelblutungen oder Blutungen in der Mundhöhle |
20 - 40 |
Alle 12 - 24 Stunden wiederholen. Mindestens 1 Tag, bis die Blutung (angezeigt durch Schmerzen) steht oder die Wundheilung erreicht ist. |
|
Schwerere Gelenks- und Muskelblutungen oder Hämatome |
30 - 60 |
Infusion alle 12 - 24 Stunden wiederholen; normalerweise 3 - 4 Tage lang oder länger, bis Schmerzen und akute Beeinträchtigung beseitigt sind. |
|
Lebensbedrohliche Blutungen |
60 - 100 |
Infusion alle 8 - 24 Stunden wiederholen, bis die Gefahr für den Patienten abgewendet ist. |
|
Chirurgische Eingriffe | ||
|
Kleinere Eingriffe einschließlich Zahnextraktion |
30 - 60 |
Alle 24 Stunden; mindestens 1 Tag, bis die Wundheilung erreicht ist. |
|
Größere Eingriffe |
80 - 100 (prä- und postoperativ) |
Die Infusion alle 8 - 24 Stunden wiederholen, bis eine adäquate Wundheilung erreicht ist; anschließend eine Behandlungsdauer von mindestens 7 weiteren Tagen, um eine Faktor VIII-Aktivität von 30 % bis 60 % (I.E./dl) aufrechtzuerhalten. |
Unter bestimmten Umständen können höhere Dosen als die hier berechneten erforderlich sein. Dies gilt insbesondere für die Anfangsdosis.
Während des Behandlungsverlaufes wird eine geeignete Bestimmung der Faktor VIII-Plasmaspiegel angeraten, um die zu verabreichende Dosis und die Häufigkeit der Injektionen zu steuern. Besonders bei größeren chirurgischen Eingriffen ist eine genaue Überwachung der Substitutionstherapie durch eine Gerinnungsanalyse (Bestimmung der Faktor VIII-Aktivität im Plasma) unerlässlich. Einzelne Patienten können sich in ihrer Reaktion auf Faktor VIII unterscheiden, verschiedene in vivo-Recovery-Werte erreichen und unterschiedliche Halbwertszeiten aufweisen.
Langzeitprophylaxe
Zur Langzeitprophylaxe von Blutungen bei Patienten mit schwerer Hämophilie A werden normalerweise Dosen zwischen 20 und 40 I.E. Faktor VIII pro kg Körpergewicht in Intervallen von 2 - 3 Tagen gegeben. In manchen Fällen, besonders bei jüngeren Patienten, können kürzere Dosierungsabstände oder höhere Dosen erforderlich sein.
B) Dosierung bei Hämophilie-Patienten mit Faktor Vlll-Inhibitoren:
Die Patienten müssen auf die Bildung von Faktor VIII-Inhibitoren überwacht werden. Falls die erwarteten Faktor VIII-Plasmaaktivitäten nicht erreicht werden oder die Blutung mit einer angemessenen Dosis nicht beherrscht wird, muss ein Faktor VIII-Inhibitortest durchgeführt werden. Bei einem Inhibitortiter von weniger als 10 B.E./ml kann der Inhibitor durch eine entsprechend höhere Dosierung von Blutgerinnungsfaktor VIII vom Menschen neutralisiert werden. Bei Patienten mit hochtitrigem Inhibitor kann die Faktor VIII-Therapie gegebenenfalls nicht effektiv sein. In diesem Fall sollen andere therapeutische Möglichkeiten in Betracht gezogen werden. Diese Therapien dürfen nur unter der Leitung von Ärzten durchgeführt werden, die mit der Behandlung von Hämophilie-Patienten vertraut sind.
C) Dosierung bei von-Willebrand-Syndrom mit Faktor VIII-Mangel:
IMMUNATE ist angezeigt für die Faktor VIII-Substitutionstherapie bei Patienten mit von-Willebrand-Syndrom, deren Faktor VIII-Aktivität reduziert ist. Die Substitutionstherapie mit IMMUNATE, um Blutungen zu kontrollieren und um Blutungen im Zusammenhang mit chirurgischen Eingriffen zu verhindern, folgt denselben Richtlinien wie für Hämophilie A.
Wird ein von-Willebrand-Faktor Produkt verwendet, das Faktor VIII enthält, so sollte dem behandelnden Arzt bewusst sein, dass bei einer längerfristigen Behandlung der Faktor-VIII:C-Spiegel stark ansteigen kann. Um einen zu hohen Faktor-VIII:C-Spiegel zu vermeiden, sollte in Betracht gezogen werden, nach einer Behandlungsdauer von 24 bis 48 Stunden die Dosis zu reduzieren und/oder die Dosisintervalle zu verlängern.
Art der Anwendung
Die Lösung herstellen, wie im Folgenden beschrieben, und langsam intravenös injizieren oder infundieren. Die Verabreichungsgeschwindigkeit sollte nicht mehr als 2 ml/Min betragen.
IMMUNATE erst unmittelbar vor der Verabreichung auflösen. Die fertige Lösung sofort verwenden (sie enthält keine Konservierungsmittel). Lösungen, die trüb sind oder Ablagerungen aufweisen, nicht verwenden. Restmengen ordnungsgemäß verwerfen.
Auflösen des Pulvers: Auf aseptische Arbeitsweise achten
1. Die ungeöffnete Lösungsmittelflasche auf Raumtemperatur erwärmen (max. 37 °C).
2. Die Schutzkappen von den Flaschen mit Pulver und Lösungsmittel entfernen (Abb. A) und die Gummistopfen beider Flaschen reinigen.
3. Transferset mit der gewellten Seite auf die Lösungsmittelflasche aufsetzen und eindrücken (Abb. B).
4. Schutzkappe von der anderen Seite des Transfersets abziehen. Freies Kanülenende nicht berühren!
5. Transferset mit aufgesetzter Wasserflasche von oben in die Flasche mit Pulver einstechen (Abb. C). Durch das in der Flasche mit Pulver bestehende Vakuum wird das Lösungsmittel angesaugt.
6. Nach etwa einer Minute Lösungsmittelflasche samt Transferset von der Konzentratflasche abziehen (Abb. D). Da sich das Präparat rasch löst, die Konzentratflasche, wenn überhaupt, nur vorsichtig schwenken. DEN INHALT DER KONZENTRATFLASCHE NICHT SCHÜTTELN! DIE KONZENTRATFLASCHE ERST UNMITTELBAR VOR DER ENTNAHME DES INHALTS UMDREHEN.
7. Parenterale Arzneimittelprodukte nach dem Auflösen und vor der Verabreichung stets visuell auf Partikel und Verfärbung überprüfen. Gelegentlich können jedoch wenige kleine Partikel sichtbar sein, auch wenn das Auflösen strikt nach Anleitung durchgeführt wurde. Das mitgelieferte Filterset entfernt diese Partikel, ohne die auf der Packung angegebene Konzentration des arzneilich wirksamen Bestandteils zu verringern.
Anwendung: Auf aseptische Arbeitsweise achten
1. Um zu verhindern, dass vom Stopfen ausgestochene Gummipartikel verabreicht werden (Gefahr von Mikroembolien) ist zur Entnahme des gelösten Präparats das beigepackte Filterset zu benutzen. Filterset auf die beigepackte Einmalspritze setzen und in den Gummistopfen einstechen (Abb. E).
2. Durch zwischenzeitliches Abziehen der Spritze vom Filterset wird die Konzentratflasche belüftet, wodurch eventuell entstandener Schaum zusammenfällt. Daraufhin die Injektionslösung durch das Filterset in die Spritze aufziehen (Abb. F).
3. Anschließend Spritze vom Filterset abziehen und die Lösung mit Hilfe des mitgelieferten Infusionssets (oder der mitgelieferten Einmalkanüle) langsam intravenös applizieren (maximale Injektionsrate: 2 ml pro Minute).
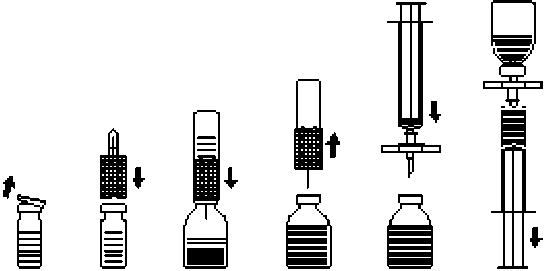
Abb.A Abb.B Abb.C Abb.D Abb. E Abb.F
Restmengen, leere Fläschchen sowie gebrauchte Kanülen und Spritzen ordnungsgemäß entsorgen.
Wenn Sie eine größere Menge von IMMUNATE angewendet haben, als Sie sollten
Es sind keine Symptome der Überdosierung mit Blutgerinnungsfaktor VIII vom Menschen bekannt.
Es besteht ein Risiko für thrombotische Ereignisse. Siehe Abschnitt 2.
Bei Patienten mit den Blutgruppen A, B oder AB besteht ein Hämolyse-Risiko. Siehe Abschnitt 2.
Wenn Sie weitere Fragen zur Anwendung dieses Arzneimittels haben, wenden Sie sich an Ihren Arzt oder Apotheker.
4. Welche Nebenwirkungen sind möglich?
Wie alle Arzneimittel kann auch dieses Arzneimittel Nebenwirkungen haben, die aber nicht bei jedem auftreten müssen.
Die unerwünschten Effekte sind in den beigefügten Listen aufgeführt, sortiert nach Berichten aus Klinischen Prüfungen und Spontanmeldungen von nicht S/D behandeltem IMMUNATE. Die Häufigkeit wurde nach den folgenden Kriterien angegeben:
Sehr häufig: (>1/10), häufig: (>1/100, <1/10), gelegentlich: (>1/1.000, <1/100), selten: (>1/10.000, <1/1.000) und sehr selten: (<1/10.000).
Klinische Prüfungen
Die Inzidenzrate ist für alle unten genannten Nebenwirkungen “gelegentlich”
GI IMMUNATE Seite 7 von 11
(>1/1.000, <1/100)
Erkrankungen des Immunsystems Allergische Reaktionen
Anwendungsbeobachtungen
Die Inzidenzrate beträgt für alle unten genannten Nebenwirkungen „sehr selten”, (d. h. <1/10.000)
Erkrankungen des Blutes und des Lymphsystems Antikörper (Inhibitoren) gegen Faktor VIII
Erkrankungen des Immunsystems Angioödem generalisierte Urtikaria Nesselsucht
Psychiatrische Erkrankungen Unruhe
Erkrankungen des Nervensystems Ameisenlaufen (Parästhesie)
Benommenheit
Kopfschmerzen
Augenerkrankungen
Lidödeme
Herzerkrankungen
Beschleunigter Herzschlag (Tachykardie)
Herzrasen
Gefäßerkrankungen
Rötung
Blutdruckabfall
Blässe
Erkrankungen der Atemwege, des Brustraums und Mediastinums
Atembeschwerden
Husten
Erkrankungen des Gastrointestinaltrakts
Übelkeit
Erbrechen
Erkrankungen der Haut und des Unterhautzellgewebes Nesselsucht
Hautausschlag (erythematös oder papulös)
Juckreiz
Hautrötung
Übermäßige Schweißproduktion
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen Muskelschmerzen
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Brennen und Stechen an der Infusionsstelle
Schüttelfrost
Teilnahmslosigkeit
Schmerzen in der Brust
Engegefühl in der Brust
Ödeme (inklusive Gesicht und periphere Ödeme)
Fieber
Die unten genannte Auflistung unerwünschter Effekte spiegelt die Art der Nebenwirkungen wider, die bei IMMUNATE auftreten können:
Erkrankungen des Blutes und des Lymphsystems Hämolysen bei Patienten mit den Blutgruppen A, B oder AB
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort Vermindertes therapeutisches Ansprechen
In einer klinischen Studie mit IMMUNATE bei 31 Patienten mit Hämophilie A wurde über eine (1) nicht schwerwiegende Nebenwirkung berichtet (leichter Schmerz an der Infusionsstelle).
In seltenen Fällen sind Überempfindlichkeits- oder allergische Reaktionen aufgetreten (z. B. angioneurotisches Ödem, brennender Schmerz an der Infusionsstelle, Schüttelfrost, Rötung, Nesselsucht, Kopfschmerzen, generalisierte Urtikaria, niedriger Blutdruck, Teilnahmslosigkeit, Übelkeit, Unruhe, Tachykardie, Spannungsgefühl in der Brust, prickelndes Gefühl, Erbrechen, Atembeschwerden, keuchende Atmung, Auftreten von Hemmkörpern gegen den von-Willebrand-Faktor). Die erforderliche Behandlung hängt von Art und Schweregrad der Reaktion ab.
In seltenen Fällen wurde ein Anstieg der Körpertemperatur beobachtet.
Patienten mit Hämophilie A können Antikörper (Inhibitoren) gegen Faktor VIII entwickeln. Wenn solche Inhibitoren auftreten, manifestiert sich dieser Zustand als eine unzureichende klinische Antwort. In diesen Fällen wird empfohlen, ein spezialisiertes Hämophilie-Zentrum zu besuchen.
Bei der Behandlung von Patienten mit von-Willebrand-Syndrom besteht ein Risiko hinsichtlich des Auftretens thrombotischer Ereignisse, besonders bei Vorliegen von bekannten klinischen oder labortechnisch belegten Risikofaktoren. Siehe auch unter Abschnitt 2.
Patienten mit von-Willebrand-Syndrom, besonders solche mit Typ 3, können neutralisierende Antikörper (Inhibitoren), gegen den von-Willebrand-Faktor entwickeln. Siehe auch unter Abschnitt 2.
Informationen zur Virussicherheit siehe unter Abschnitt 2.
Meldung von Nebenwirkungen
Wenn Sie Nebenwirkungen bemerken, wenden Sie sich an Ihren Arzt oder das medizinische Fachpersonal. Dies gilt auch für Nebenwirkungen, die nicht in dieser Packungsbeilage angegeben sind. Sie können Nebenwirkungen auch direkt dem Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel, Paul-Ehrlich-Institut, Paul-Ehrlich-Str. 51-59, 63225 Langen, Tel: +49 6103 77 0, Fax: +49 6103 77 1234,
Website: www.pei.de anzeigen. Indem Sie Nebenwirkungen melden, können Sie dazu beitragen, dass mehr Informationen über die Sicherheit dieses Arzneimittels zur Verfügung gestellt werden.
5. Wie ist IMMUNATE aufzubewahren?
Bewahren Sie dieses Arzneimittel für Kinder unzugänglich auf.
Sie dürfen dieses Arzneimittel nach dem auf dem Etikett angegebenen Verfalldatum nicht mehr verwenden. Das Verfalldatum bezieht sich auf den letzten Tag des angegebenen Monats.
Die chemische und physikalische Stabilität der gebrauchsfertigen Lösung wurde für 3 Stunden bei Raumtemperatur nachgewiesen. Aus mikrobiologischer Sicht sollte die Lösung dennoch unmittelbar verwendet werden, außer die Herstellung der gebrauchsfertigen Lösung schließt das Risiko einer mikrobiologischen Kontamination aus. Wird die gebrauchsfertige Lösung nicht unverzüglich verwendet, liegen Lagerbedingungen und -zeit in der Verantwortung des Anwenders. Gebrauchsfertige Lösung darf nicht in den Kühlschrank zurückgestellt werden
Besondere Lagerungshinweise
IMMUNATE zwischen +2 °C und +8 °C (im Kühlschrank) lagern.
In der Originalpackung aufbewahren, um das Produkt vor Licht zu schützen.
Entsorgen Sie Arzneimittel nicht im Abwasser oder Haushaltsabfall. Fragen Sie Ihren Apotheker, wie das Arzneimittel zu entsorgen ist, wenn Sie es nicht mehr verwenden. Sie tragen damit zum Schutz der Umwelt bei.
6. Inhalt der Packung und weitere Informationen Was IMMUNATE enthält
Der Wirkstoff ist Blutgerinnungsfaktor VIII vom Menschen.
1 Durchstechflasche Pulver enthält:
|
IMMUNATE STIM plus Immuno |
250 I.E. |
500 I.E. |
1000 I.E. |
|
Blutgerinnungsfaktor VIII vom Menschen |
250 I.E* |
500 I.E* |
1000 I.E* |
|
Proteingehalt (Faktor VIII) |
2,5 - 6,3 mg |
5 - 12,5 mg |
10 - 25 mg |
|
Spezifische Aktivität |
70 ± 30 I.E./mg Protein** | ||
|
von-Willebrand-Faktor (VWF:RCo) vom Menschen |
150 I.E.*** |
300 I.E.*** |
600 I.E.*** |
Nach Auflösen mit 5 ml (250 I.E. und 500 I.E. Packungsgröße) oder 10 ml (1000 I.E. Packungsgröße) sterilisiertem Wasser für Injektionszwecke enthält das Produkt 50 I.E./ml (250 I.E. Packungsgröße) oder 100 I.E./ml (500 I.E. und 1000 I.E. Packungsgröße) an Blutgerinnungsfaktor VIII vom Menschen und 30
GI IMMUNATE Seite 10 von 11
I.E./ml (250 I.E. Packungsgröße) oder 60 I.E./ml (500 I.E. und 1000 I.E. Packungsgröße) an von-Willebrand-Faktor vom Menschen.
*) Die Faktor VIII-Aktivität wurde gegen den internationalen Standard der WHO für Faktor VIII-Konzentrate bestimmt.
**) ohne Stabilisator (Albumin)
Die maximale spezifische Aktivität bei einem Verhältnis von 1:1 zwischen der Faktor VIII-Aktivität und von-Willebrand-Faktor-Antigen beträgt 100 I.E. Faktor VIII pro mg Protein.
***) Die Ristocetin-Cofaktor-Aktivität von humanem von-Willebrand-Faktor bezieht sich auf den Internationalen Standard der WHO für von-Willebrand-Faktor -Konzentrate.
Die sonstigen Bestandteile sind:
Pulver:
Albumin, Glycin, Lysinhydrochlorid, Natriumchlorid, Natriumcitrat, Calciumchlorid Lösungsmittel:
Sterilisiertes Wasser für Injektionszwecke
Wie IMMUNATE aussieht und Inhalt der Packung
Pulver und Lösungsmittel zur Herstellung einer Infusionslösung
IMMUNATE ist in folgenden Größen erhältlich: 250 I.E, 500 I.E. und 1000 I.E., aufzulösen in 5 ml (250 I.E. und 500 I.E.) bzw. 10 ml (1000 I.E.) Sterilisiertem Wasser für Injektionszwecke. Sowohl Pulver als auch Lösungsmittel sind in Einzeldosis-Glasfläschchen mit Gummi-Stopfen abgefüllt. Jede Packung enthält auch ein Set zum Auflösen und Verabreichen.
Pharmazeutischer Unternehmer und Hersteller
Pharmazeutischer Unternehmer
Baxter Deutschland GmbH
Edisonstraße 4
85716 Unterschleißheim
Hersteller
Baxter AG Industriestraße 67 A-1221 Wien
Herkunftsländer der zur Produktion verwendeten Plasmen
Deutschland, Finnland, Norwegen, Österreich, Schweden, Schweiz, Tschechien und Vereinigte Staaten von Amerika.
Diese Packungsbeilage wurde zuletzt überarbeitet im Dezember 2014.
GI IMMUNATE Seite 11 von 11