Immunate Stim Plus 250 I.e. Immuno
alt informationenFACHINFORMATION
1. BEZEICHNUNG DES ARZNEIMITTELS
IMMUNATE STIM plus 250 I.E./500 I.E./1000 I.E. Immuno Pulver und Lösungsmittel zur Herstellung einer Injektionslösung
2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
1 Durchstechflasche Pulver enthält:
|
IMMUNATE STIM plus Immuno |
250 I.E. |
500 I.E. |
1000 I.E. |
|
Blutgerinnungsfaktor VIII vom Menschen |
250 I.E* |
500 I.E* |
1000 I.E* |
|
Proteingehalt (Faktor VIII) |
2,5 - 6,3 mg |
5 - 12,5 mg |
10 - 25 mg |
|
Spezifische Aktivität |
70 ± 30 I.E./mg Protein** | ||
|
Von-Willebrand-Faktor (vWF:RCo) vom Menschen |
150 I.E.*** |
300 I.E.*** |
600 I.E.*** |
Nach Auflösen mit 5 ml (250 I.E. und 500 I.E. Packungsgröße) oder 10 ml (1000 I.E. Packungsgröße) sterilisiertem Wasser für Injektionszwecke enthält das Produkt
50 I.E./ml (250 I.E. Packungsgröße) oder 100 I.E./ml (500 I.E. und 1000 I.E. Packungsgröße) an Blutgerinnungsfaktor VIII vom Menschen und 30 I.E./ml (250 I.E. Packungsgröße) oder 60 I.E./ml (500 I.E. und 1000 I.E. Packungsgröße) an von-Willebrand-Faktor vom Menschen.
*) Die FVIII-Konzentrationsangabe bezieht sich auf den Internationalen Standard der Weltgesundheitsorganisation WHO für Faktor VIII-Konzentrate.
**) ohne Stabilisator (Albumin)
Die maximale spezifische Aktivität bei einem Verhältnis von 1:1 zwischen der Faktor VIII-Aktivität und von-Willebrand-Faktor-Antigen beträgt 100 I.E. Faktor VIII pro mg Protein.
***) Die Ristocetin-Cofaktor-Aktivität von humanem von-Willebrand-Faktor bezieht sich auf den Internationalen Standard der Weltgesundheitsorganisation WHO für von-Willebrand-Faktor-Konzentrat.
Sonstige Bestandteile mit bekannter Wirkung: Eine Durchstechflasche enthält ca. 9,8 mg Natrium (250 I.E. und 500 I.E. Packungsgröße) bzw. 19,6 mg Natrium (1000 I.E. Packungsgröße).
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
3. DARREICHUNGSFORM
Pulver und Lösungsmittel zur Herstellung einer Injektionslösung.
4. KLINISCHE ANGABEN
4.1 Anwendungsgebiete
Behandlung und Prophylaxe von Blutungen bei Patienten mit angeborenem oder erworbenem Faktor VIII-Mangel (Hämophilie A, Hämophilie A mit Faktor VIII-Inhibitor, erworbener Faktor VIII-Mangel aufgrund einer spontanen Entwicklung von Faktor VIII-Inhibitor).
Behandlung von Blutungen bei Patienten mit von-Willebrand-Syndrom mit Faktor VIII-Mangel, wenn kein spezifisches bei von-Willebrand-Syndrom wirksames Plasmapräparat zur Verfügung steht.
4.2 Dosierung und Art der Anwendung
Die Behandlung ist unter Überwachung eines Arztes einzuleiten, der auf dem Gebiet der Hämophiliebehandlung erfahren ist.
Dosierung
A. Dosierung bei Hämophilie A:
Dosierung und Dauer der Substitutionstherapie hängen von der Schwere des Faktor VIII-Mangels sowie dem Ort und Ausmaß der Blutung und dem klinischen Zustand des Patienten ab.
Die Anzahl der verabreichten Faktor VIII-Einheiten ist in Internationalen Einheiten (I.E.) angegeben, die sich auf den aktuellen Standard der WHO für Faktor VIII-Produkte beziehen. Die Faktor VIII-Aktivität im Plasma wird entweder prozentual (relativ zu normalem Humanplasma) oder in Internationalen Einheiten (bezogen auf den Internationalen Standard für Faktor VIII-Konzentrate) ausgedrückt.
Eine Internationale Einheit (I.E.) Faktor VIII-Aktivität entspricht der Menge an Faktor VIII in 1 ml normalem Humanplasma.
Die unten angegebene Berechnung der erforderlichen Dosis Faktor VIII beruht auf der empirischen Erkenntnis, dass 1 Internationale Einheit (I.E.) Faktor VIII pro kg Körpergewicht die Faktor VIII-Aktivität im Plasma um ca. 2 % der normalen Aktivität erhöht.
Die erforderliche Dosis wird mit folgender Formel ermittelt:
Erforderliche Einheiten = Körpergewicht (kg) x gewünschter Faktor-VIII-Anstieg (%) x 0,5
Die Dosis und das Dosierungsintervall sollten sich immer nach der klinischen Wirksamkeit im Einzelfall richten.
Blutungen und chirurgische Eingriffe
Bei folgenden Blutungsereignissen darf die Faktor VIII-Aktivität im entsprechenden Zeitraum nicht unter die angegebenen Plasmaspiegel sinken.
Die folgende Tabelle enthält Richtwerte für die Dosierung bei Blutungen und chirurgischen Eingriffen:
|
Grad der Blutung/Art des chirurgischen Eingriffs |
derlicher FVIII el (% des alwerts) (I.E./dl) |
Substitutionsintervall (Stunden) / Behandlungsdauer (Tage) |
|
Blutung | ||
|
Frühstadium von Gelenks- und Muskelblutungen oder Blutungen in der Mundhöhle |
0 |
Alle 12 - 24 Stunden wiederholen. Mindestens 1 Tag, bis die Blutung (angezeigt durch Schmerzen) steht oder die Wundheilung erreicht ist. |
|
Schwerere Gelenks- und Muskelblutungen oder Hämatome |
0 |
Infusion alle 12 - 24 Stunden wiederholen; normalerweise 3 -4 Tage lang oder länger, bis Schmerzen und akute Beeinträchtigung beseitigt sind. |
|
Lebensbedrohliche Blutungen |
00 |
Infusion alle 8 - 24 Stunden wiederholen, bis die Gefahr für den Patienten abgewendet ist. |
|
Chirurgische Eingriffe | ||
|
Kleinere Eingriffe einschließlich Zahnextraktion |
0 |
Alle 24 Stunden; mindestens 1 Tag, bis die Wundheilung erreicht ist. |
|
Größere Eingriffe |
80 - 100 (prä- und postoperativ) |
Die Infusion alle 8 - 24 Stunden wiederholen, bis eine adäquate Wundheilung erreicht ist; anschließend eine Behandlungsdauer von mindestens 7 weiteren Tagen, um eine Faktor-VIII-Aktivität von 30 % bis 60 % (I.E./dl) aufrechtzuerhalten. |
Unter bestimmten Umständen können höhere Dosen als die hier berechneten erforderlich sein. Dies gilt insbesondere für die Anfangsdosis.
Während des Behandlungsverlaufes wird eine angemessene Bestimmung der Faktor-VIII-Plasmaspiegel angeraten, um die zu verabreichende Dosis und die Häufigkeit der Injektionen zu steuern. Besonders bei größeren chirurgischen Eingriffen ist eine genaue Überwachung der Substitutionstherapie durch eine Gerinnungsanalyse (Bestimmung der Faktor-VIII-Aktivität im Plasma) unerlässlich. Einzelne Patienten können sich in ihrer Reaktion auf Faktor VIII unterscheiden, verschiedene in-vivo-Recovery-Werte erreichen und unterschiedliche Halbwertszeiten aufweisen.
Langzeitprophylaxe
Zur Langzeitprophylaxe von Blutungen bei Patienten mit schwerer Hämophilie A werden normalerweise Dosen zwischen 20 und 40 I.E. Faktor VIII pro kg Körpergewicht in Intervallen von 2 - 3 Tagen gegeben. In manchen Fällen, besonders bei jüngeren Patienten, können kürzere Dosierungsabstände oder höhere Dosen erforderlich sein.
B. Hämophilie-Patienten mit Faktor-VIII-Inhibitoren:
FI IMMUNATE
Die Patienten müssen auf die Bildung von Faktor VIII-Inhibitoren überwacht werden. Falls die erwarteten Faktor-VIII-Plasmaaktivitäten nicht erreicht werden oder die Blutung mit einer angemessenen Dosis nicht beherrscht wird, muss ein Faktor-VIII-Inhibitortest durchgeführt werden. Bei einem Inhibitortiter von weniger als 10 B.E./ml kann der Inhibitor durch eine entsprechend höhere Dosierung von Blutgerinnungsfaktor VIII vom Menschen neutralisiert werden. Bei Patienten mit hochtitrigem Inhibitor kann die Faktor VIII-Therapie gegebenenfalls nicht effektiv sein. In diesem Fall sollen andere therapeutische Möglichkeiten in Betracht gezogen werden. Diese Therapien dürfen nur unter der Leitung von Ärzten durchgeführt werden, die mit der Behandlung von Hämophilie-Patienten vertraut sind.
Siehe auch 4.4.
C. Von-Willebrand-Syndrom mit Faktor VIII-Mangel:
IMMUNATE ist angezeigt für die Faktor-VIII-Substitutionstherapie bei Patienten mit von-Willebrand-Syndrom, deren Faktor-VIII-Aktivität reduziert ist. Die Substitutionstherapie mit IMMUNATE, um Blutungen zu kontrollieren und um Blutungen im Zusammenhang mit chirurgischen Eingriffen zu verhindern, folgt denselben Richtlinien wie für Hämophilie A.
Wird ein von-Willebrand-Faktor Produkt verwendet, das Faktor VIII enthält, so sollte dem behandelnden Arzt bewusst sein, dass bei einer längerfristigen Behandlung der Faktor-VIII:C-Spiegel stark ansteigen kann. Um einen zu hohen Faktor-VIII:C-Spiegel zu vermeiden, sollte in Betracht gezogen werden, nach einer Behandlungsdauer von 24 bis 48 Stunden die Dosis zu reduzieren und/oder die Dosisintervalle zu verlängern.
Art der Anwendung
Die Lösung herstellen, wie in Abschnitt 6.6 beschrieben, und langsam intravenös injizieren oder infundieren. Es wird empfohlen, nicht mehr als 2 ml pro Minute zu verabreichen.
4.3 Gegenanzeigen
Überempfindlichkeit gegenüber einem der arzneilich wirksamen Bestandteile oder einem der sonstigen Bestandteile.
4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Wenn während der Verabreichung von IMMUNATE Überempfindlichkeitsreaktionen auftreten, ist die Injektion/Infusion abzubrechen und eine entsprechende medizinische Behandlung einzuleiten. Die Patienten müssen deshalb über frühe Anzeichen von Überempfindlichkeitsreaktionen aufgeklärt werden, wie z. B. Nesselsucht, generalisierte Urtikaria, Tachykardie, Schmerzen in der Brust, Atembeschwerden, Ödeme (inklusive Gesichts- und Lidödeme), Hautausschlag, Gesichtsrötung, Pruritus, niedriger Blutdruck sowie Anaphylaxie bis hin zum anaphylaktischen Schock. Die aktuellen medizinischen Richtlinien zur Schocktherapie sind zu beachten.
Standardmaßnahmen zur Vorbeugung von Infektionen, die sich durch den Einsatz von Arzneimitteln ergeben, die aus Blut oder Blutplasma hergestellt sind, schließen die Auswahl der Spender und das Screening der einzelnen Blutspenden und Plasmapools auf spezifische Infektionsmarker sowie effektive Schritte zur Inaktivierung/Eliminierung von Viren im Herstellungsverfahren ein. Dennoch kann bei der Verabreichung von Arzneimitteln aus menschlichem Blut oder Blutplasma die Möglichkeit der Übertragung von Krankheitserregern nicht völlig ausgeschlossen werden. Dasselbe gilt auch für bislang unbekannte oder neu aufgetretene Viren und andere Pathogene.
Die durchgeführten Maßnahmen werden als wirksam gegen umhüllte Viren wie HIV, HBV und HCV und gegen das nicht-umhüllte Virus HAV betrachtet. Gegen nicht-umhüllte Viren wie das Parvovirus B19 können diese Maßnahmen möglicherweise nur begrenzt wirksam sein.
Parvovirus B19-Infektionen können schwerwiegende Folgen bei Schwangeren haben (Infektion des Fötus) und bei Menschen, die an einem Immundefekt oder einem erhöhten Erythrozytenabbau (z. B. hämolytische Anämie) leiden.
Für Patienten, die regelmäßig/oder wiederholt aus Blutplasma hergestellte Faktor VIII-Konzentrate/von-Willebrand-Faktoren-Konzentrate, wie IMMUNATE erhalten, sollte ein geeigneter Impfschutz (Hepatitis A und B) in Betracht gezogen werden.
Es wird dringend empfohlen, dass bei jeder Verabreichung von IMMUNATE Name und Chargenbezeichnung des Präparates dokumentiert werden, um eine Rückverfolgung vom Patient zur Produktcharge sicherstellen zu können.
Bei der Behandlung von Patienten mit von-Willebrand-Syndrom besteht ein Risiko hinsichtlich des Auftretens thrombotischer Ereignisse, besonders bei Vorliegen von bekannten klinischen oder labortechnisch belegten Risikofaktoren. Deshalb müssen Risikopatienten auf frühe Zeichen von Thrombosen hin überwacht werden. Eine Prophylaxe gegenüber Thromboembolien sollte gemäß den aktuellen Empfehlungen durchgeführt werden. Bei Patienten, die mit einem von-Willebrand-Faktor Produkt behandelt werden, das Faktor VIII enthält, kann der Faktor-VIII:C-Spiegel stark ansteigen. Es sollte daher unbedingt der Faktor-VIII:C-Plasmaspiegel überwacht werden, um eine dauerhafte Erhöhung zu vermeiden, da dies mit einem erhöhten Risiko für thrombotische Ereignisse einhergeht.
Bei Patienten, bei denen in der Vergangenheit bereits venöse Thromboembolien aufgetreten sind, wurde ein endogener hoher Faktor-VIII-Spiegel mit einem erhöhten Risiko für spätere thrombotische Ereignisse in Verbindung gebracht.
Patienten mit von-Willebrand-Syndrom, besonders solche mit Typ 3, können neutralisierende Antikörper (Inhibitoren), gegen den von-Willebrand-Faktor entwickeln. Wenn der erwartete Anstieg der von-Willebrand-Ristocetin-Cofaktor-Aktivität im Plasma nicht erreicht wird, oder wenn Blutungen nicht mit einer entsprechenden Dosis beherrscht werden können, sollten geeignete Tests auf die Anwesenheit von von-Willebrand-Inhibitoren durchgeführt werden. Es besteht die Möglichkeit, dass bei Patienten mit hohen Inhibitortitern die von-Willebrand-Therapie nicht effektiv ist und andere therapeutische Möglichkeiten in Betracht gezogen werden sollten. Die Behandlung solcher Patienten sollte unter der Leitung von Ärzten durchgeführt werden, die mit der Behandlung von Patienten mit Gerinnungsstörungen vertraut sind.
Diese Antikörper können auch eine Anaphylaxie auslösen. Patienten, bei denen es zu einer anaphylaktischen Reaktion kommt, sollten daher auf Inhibitoren gegen den von-Willebrand-Faktor untersucht werden.
Die Bildung von Antikörpern mit Faktor-VIII-neutralisierender Wirkung (Inhibitoren) ist eine bekannte Komplikation bei der Behandlung von Hämophilie A-Patienten. Diese Inhibitoren sind meist IgG-Immunglobuline, deren inhibitorische Aktivität gegen Faktor VIII in Bethesda-Einheiten (B.E.) pro ml Plasma (modifizierter Bethesda Assay) ausgedrückt wird. Das Risiko einer Hemmkörper-Entwicklung wird von einer Reihe von Faktoren bestimmt, die mit speziellen Eigenschaften des jeweiligen Patienten zusammenhängen. Als wichtigste Risikofaktoren gelten unter anderem die Art der FVIII-Genmutation, die Familienanamnese und die ethnische Zugehörigkeit.
Über Inhibitoren wurden überwiegend bei nicht vorbehandelten Patienten berichtet.
Das Risiko einer Bildung von Inhibitoren hängt mit der Exposition gegenüber Antihämophilie-Faktor VIII zusammen, wobei dieses Risiko innerhalb der ersten 20 Expositionstage am höchsten ist. In seltenen Fällen können sich Inhibitoren auch nach den ersten 100 Expositionstagen bilden.
Patienten, die mit Blutgerinnungsfaktor VIII vom Menschen behandelt wurden, sind mit entsprechenden klinischen Überwachungsmethoden und Laboruntersuchungen sorgfältig auf die Bildung von inhibitorischen Antikörpern zu überwachen. Siehe auch 4.8.
Das Produkt ist bei Kindern unter sechs Jahren, die noch nicht häufig mit Faktor VIII-Produkten behandelt wurden, mit besonderer Vorsicht anzuwenden, da systematische klinische Untersuchungen an Kindern nicht durchgeführt wurden.
IMMUNATE enthält Blutgruppen-Isoagglutinine (anti-A und anti-B). Bei Patienten mit den Blutgruppen A, B oder AB kann es nach wiederholter Anwendung in kurzen Abständen oder nach der Anwendung sehr hoher Dosen zu einer Hämolyse kommen. Sehr hohe Dosen innerhalb kurzer Zeit werden möglicherweise im Rahmen einer Immuntoleranztherapie zur Behandlung einer Hämophilie A mit Faktor-VIII-Inhibitor eingesetzt.
Bei Patienten, die auf eine natriumarme Ernährung achten müssen, muss der Natriumgehalt berücksichtigt werden, da der Natriumgehalt in der maximalen täglichen Dosis eventuell höher ist als 200 mg.
4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Es wurden keine Studien zur Erfassung von Wechselwirkungen durchgeführt.
Derzeit sind keine pharmakologischen Wechselwirkungen mit anderen Arzneimitteln bekannt.
4.6 Fertilität, Schwangerschaft und Stillzeit
Reproduktionstoxikologische Studien mit Labortieren wurden nicht durchgeführt. Die Unbedenklichkeit von Blutgerinnungsfaktor VIII vom Menschen bei Schwangerschaften wurde nicht in kontrollierten klinischen Versuchen festgestellt. IMMUNATE ist daher während der Schwangerschaft und Stillzeit nur anzuwenden, wenn dies unbedingt erforderlich ist.
Informationen zu Parvovirus B19-Infektionen siehe Abschnitt 4.4.
4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Es sind keine Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen bekannt.
4.8 Nebenwirkungen
Die unerwünschten Effekte sind in den beigefügten Listen aufgeführt, sortiert nach Berichten aus Klinischen Prüfungen und Spontanmeldungen von nicht-S/D-behandeltem IMMUNATE. Die Häufigkeit wurde nach den folgenden Kriterien angegeben:
Sehr häufig: (>1/10), häufig: (>1/100, <1/10), gelegentlich: (>1/1.000, <1/100), selten: (>1/10.000, <1/1.000), sehr selten: (<1/10.000) und nicht bekannt: (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
Klinische Prüfungen:
Die Häufigkeit ist für alle unten genannten Nebenwirkungen “gelegentlich”
(>1/1.000, <1/100)
Erkrankungen des Immunsystems Allergische Reaktionen
Anwendungsbeobachtungen:
Die Häufigkeit für alle unten genannten Nebenwirkungen ist nicht bekannt.
Erkrankungen des Blutes und des Lymphsystems Antikörper (Inhibitoren) gegen Faktor VIII
Erkrankungen des Immunsystems Angioödem generalisierte Urtikaria Nesselsucht
Psychiatrische Erkrankungen Unruhe
Erkrankungen des Nervensystems Parästhesie (prickelndes Gefühl)
Benommenheit
Kopfschmerzen
Augenerkrankungen
Lidödeme
Herzerkrankungen
Beschleunigter Herzschlag (Tachykardie)
Herzrasen
Gefäßerkrankungen
Rötung
Blutdruckabfall
Blässe
Erkrankungen der Atemwege, des Brustraums und des Mediastinums
Atembeschwerden
Husten
Erkrankungen des Gastrointestinaltrakts
Übelkeit
Erbrechen
Erkrankungen der Haut und des Unterhautzellgewebes Nesselsucht
Hautausschlag (erythematös oder papulös)
Juckreiz
Erythem
Hyperhidrose
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen Myalgie
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Brennen und Stechen an der Infusionsstelle
Schüttelfrost
Teilnahmslosigkeit
Schmerzen in der Brust
Engegefühl in der Brust
Ödeme (inklusive Gesicht und periphere Ödeme)
Fieber
Die unten genannte Auflistung unerwünschter Effekte spiegelt die Art der Nebenwirkungen wider, die bei IMMUNATE auftreten können:
Erkrankungen des Blutes und des Lymphsystems
Hämolysen bei Patienten mit den Blutgruppen A, B oder AB
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort Vermindertes therapeutisches Ansprechen
In einer klinischen Studie mit S/D-behandeltem IMMUNATE bei 31 Patienten mit Hämophilie A wurde über eine (1) nicht schwerwiegende Nebenwirkung berichtet (leichter Schmerz an der Infusionsstelle).
In seltenen Fällen sind Überempfindlichkeits- oder allergische Reaktionen aufgetreten (z. B. angioneurotisches Ödem, brennender Schmerz an der Infusionsstelle, Schüttelfrost, Rötung, Nesselsucht, Kopfschmerzen, generalisierte Urtikaria, niedriger Blutdruck, Teilnahmslosigkeit, Übelkeit, Unruhe, Tachykardie, Spannungsgefühl in der Brust, prickelndes Gefühl, Erbrechen, Atembeschwerden, keuchende Atmung, Inhibition des von-Willebrand-Faktors). Die erforderliche Behandlung hängt von Art und Schweregrad der Reaktion ab.
In seltenen Fällen wurde ein Anstieg der Körpertemperatur beobachtet.
Patienten mit Hämophilie A können Antikörper (Inhibitoren) gegen Faktor VIII entwickeln. Wenn solche Inhibitoren auftreten, manifestiert sich dieser Zustand als eine unzureichende klinische Antwort. In diesen Fällen wird empfohlen, ein spezialisiertes Hämophilie-Zentrum zu besuchen.
Bei der Behandlung von Patienten mit von-Willebrand-Syndrom besteht ein Risiko hinsichtlich des Auftretens thrombotischer Ereignisse, besonders bei Vorliegen von bekannten klinischen oder labortechnisch belegten Risikofaktoren. Siehe Abschnitt 4.4.
Patienten mit von-Willebrand-Syndrom, besonders solche mit Typ 3, können neutralisierende Antikörper (Inhibitoren), gegen den von-Willebrand-Faktor entwickeln. Siehe Abschnitt 4.4.
Informationen zur Virussicherheit, siehe Abschnitt 4.4.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel, Paul-Ehrlich-Institut, Paul-Ehrlich-Str. 51-59, 63225 Langen, Tel: +49 6103 77 0, Fax: +49 6103 77 1234, Website: www.pei.de anzuzeigen.
4.9 Überdosierung
Es sind keine Symptome der Überdosierung mit Blutgerinnungsfaktor VIII vom Menschen bekannt. Es besteht ein Risiko für thrombotische Ereignisse. Siehe Abschnitt 4.4.
Bei Patienten mit den Blutgruppen A, B oder AB besteht ein Hämolyse-Risiko. Siehe Abschnitt 4.4.
5. PHARMAKOLOGISCHE EIGENSCHAFTEN
5.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Antihämorrhagika, Blutgerinnungsfaktoren: von-Willebrand-Faktor human und Gerinnungsfaktor VIII in Kombination.
ATC-Code: B02BD06
Der Faktor VIII/von-Willebrand-Faktor-Komplex besteht aus zwei Molekülen (FVIII und vWF) mit unterschiedlichen physiologischen Funktionen.
Aktivierter Faktor VIII wirkt als Cofaktor für den aktivierten Faktor IX und beschleunigt die Umwandlung von Faktor X zu aktiviertem Faktor X. Der aktivierte Faktor X wandelt Prothrombin in Thrombin um. Thrombin setzt dann Fibrin aus Fibrinogen frei, und ein Blutgerinnsel kann sich bilden. Hämophilie A ist eine geschlechtsgebundene erbliche Störung der Blutgerinnung aufgrund erniedrigter Faktor VIII:C-Spiegel, die entweder spontan oder in Folge unfallbedingter oder chirurgischer Traumata zu starken Blutungen in Gelenken, Muskeln oder inneren Organen führt. Die Faktor VIII-Plasmaspiegel werden durch die Substitutionstherapie erhöht, wodurch eine vorübergehende Korrektur des Faktor-VIII-Mangels und der Blutungsneigung erzielt wird.
Zusätzlich zu seiner Aufgabe als FVIII-schützendes Protein vermittelt vWF die Thrombozytenadhäsion an Gefäßverletzungen, spielt eine Rolle bei der Thrombozytenaggregation und ist unverzichtbar für die Substitutionstherapie bei Patienten mit von-Willebrand-Syndrom:
o Der von-Willebrand-Faktor stellt im Falle einer Gefäßverletzung das Anhaften der Plättchen an das Gefäßendothel wieder her (da er sowohl an das Gefäßendothel als auch an die Plättchenmembran bindet). Dadurch wird die primäre Blutstillung erzeugt, was sich in der Verkürzung der Blutungszeit zeigt. Dieser Effekt tritt sofort ein und hängt bekanntlich in großem Ausmaß vom Polymerisationsgrad des Proteins ab.
o Der von-Willebrand-Faktor schützt den Faktor VIII vor einem vorzeitigen Abbau. Von-Willebrand-Faktor bindet an endogenen Faktor VIII (der normalerweise vom Patienten produziert wird) und, indem er dadurch diesen Faktor stabilisiert, wird dessen rascher Abbau verhindert. Die Verabreichung eines Faktor VIII-haltigen Präparates hebt den Faktor VIII-Spiegel sofort nach der ersten Infusion auf normale Plasmaspiegel an.
5.2 Pharmakokinetische Eigenschaften
Eine pharmakokinetische Studie mit 18 Patienten zeigte nach vollständiger Analyse die unten beschriebenen Ergebnisse.
Die folgende Tabelle beschreibt die pharmakokinetischen Eigenschaften des Blutgerinnungsfaktor VIII:
|
eter | |||||
|
Anzahl |
Mittelwert |
SD |
Median |
90 % CI | |
|
AUCc_48h ([I.E.xh]/ml) |
18 |
11,4 |
2,8 |
11,6 |
10,9 - 12,7 |
|
AUCq_k ([I.E.xh]/ml) |
18 |
12,2 |
3,1 |
12,4 |
11,1 - 13,2 |
|
Cmax (I.E./ml) |
18 |
1,0 |
0,3 |
0,9 |
0,8 - 1,0 |
|
Tmax (h) |
18 |
0,3 |
0,1 |
0,3 |
0,3 - 0,3 |
|
Terminale Halbwertszeit (h) |
18 |
12,7 |
3,2 |
12,2 |
10,8 - 15,3 |
|
Clearance (ml/h) |
18 |
283 |
146 |
232 |
199 - 254 |
|
Mittlere Verweilzeit (h) |
18 |
15,3 |
3,6 |
15,3 |
12,1 - 17,2 |
|
Vss (ml) |
18 |
4166 |
2021 |
3613 |
2815 - 4034 |
|
Incremental Recovery ([I.E./ml]/[I.E./kg]) |
18 |
0,020 |
0,006 |
0,019 |
0,016 - 0,020 |
In einem Teil der Hämophilie A Patienten wurden die pharmakokinetischen Eigenschaften der von-Willebrand-Komponente des Faktor VIII-von-Willebrand-Komplex untersucht.
Die folgende Tabelle beschreibt die pharmakokinetischen Eigenschaften des vWF:RCo:
|
Parameter | |||
|
Anzahl |
Median |
95 % CI | |
|
AUCq.« ([I.E. x h]/ml) Cmax (I.E./ml) Tmax (h) Terminale Halbwertszeit (h) Clearance (ml/h) Mittlere Verweilzeit (h) Vss (ml) Incremental Recovery ([I.E./ml]/[I.E./kg]) |
11 |
12,4 |
5,8 - 52,0 |
|
15 |
0,62 |
0,53 - 0,74 | |
|
15 |
0,25 |
0,25 - 0,50 | |
|
12 |
15,9 |
11,5 - 45,5 | |
|
11 |
283 |
46 - 629 | |
|
11 |
21,8 |
14,8 -141,2 | |
|
11 |
7525 |
5135 - 9377 | |
|
15 |
0,012 |
0,011 - 0,015 | |
Die folgende Tabelle beschreibt die pharmakokinetischen Eigenschaften des vWF:Ag:
|
Parameter | |||
|
Anzahl |
Median |
95 % CI | |
|
AUCq.« ([I.E. x h]/ml) Cmax (I.E./ml) Tmax (h) Terminale Halbwertszeit (h) Clearance (ml/h) Mittlere Verweilzeit (h) Vss (ml) Incremental Recovery ([I.E./ml]/[I.E./kg]) |
15 |
24,6 |
12,8 - 48,3 |
|
17 |
1,40 |
1,15 - 1,51 | |
|
17 |
0,28 |
0,25 - 1,00 | |
|
16 |
13,6 |
10,5 - 47,2 | |
|
15 |
136 |
68 - 178 | |
|
15 |
23,1 |
12,4 - 57,1 | |
|
15 |
3156 |
2391 - 4672 | |
|
17 |
0,028 |
0,024 - 0,030 | |
5.3 Präklinische Daten zur Sicherheit
Der in IMMUNATE enthaltene Blutgerinnungsfaktor VIII vom Menschen ist ein normaler Bestandteil von menschlichem Plasma und verhält sich wie körpereigener Faktor VIII.
Die einmalige Verabreichung der mehrfachen für den Menschen empfohlenen Dosis pro Kilogramm Körpergewicht gab keine Hinweise auf toxische Auswirkungen auf die Labortiere.
Tierversuche mit wiederholten Gaben sind aufgrund der Wechselwirkung durch die sich bildenden Antikörper auf Fremdprotein nicht sinnvoll durchführbar.
Die bislang vorliegenden klinischen Erfahrungen geben keinen Hinweis auf kanzerogene und mutagene Wirkungen.
Toxikologische Untersuchungen mit den Solvent/Detergent-Substanzen gaben bei Labortieren keine Hinweise auf ein erhöhtes pharmakologisch-toxikologisches Potential.
6. PHARMAZEUTISCHE ANGABEN
6.1 Liste der sonstigen Bestandteile
Albumin, Glycin, Lysinhydrochlorid, Natriumchlorid, Natriumcitrat, Calciumchlorid
6.2 Inkompatibilitäten
Wie alle Gerinnungsfaktor-Konzentrate darf auch IMMUNATE vor der Verabreichung nicht mit anderen Arzneimitteln gemischt werden, da dies die Wirksamkeit und Sicherheit des Produktes beeinträchtigen könnte. Es ist ratsam, einen gemeinsamen Venenzugang vor und nach der Infusion von IMMUNATE mit isotonischer Kochsalzlösung zu spülen.
6.3 Dauer der Haltbarkeit
2 Jahre
IMMUNATE STIM plus Immuno nach dem auf der Verpackung angegebenen Verfalldatum nicht mehr verwenden.
Die chemische und physikalische Stabilität von gelöstem IMMUNATE während der Anwendung wurde über 3 Stunden bei Raumtemperatur nachgewiesen. Unter mikrobiologischen Gesichtspunkten ist das Produkt unmittelbar zu verwenden, es sei denn, durch das Auflösungsverfahren ist eine mikrobielle Kontamination ausgeschlossen. Wird es nicht sofort verwendet, liegen die Lagerungsdauer und die Lagerungsbedingungen während der Anwendung im Verantwortungsbereich des Anwenders. Wenn das Produkt einmal aufgelöst wurde, auf keinen Fall wieder in den Kühlschrank zurückstellen.
6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
IMMUNATE zwischen +2 °C und +8 °C (im Kühlschrank) lagern.
In der Originalpackung aufbewahren, um das Produkt vor Licht zu schützen.
Aufbewahrungsbedingungen nach Rekonstitution des Arzneimittels, siehe Abschnitt 6.3.
6.5 Art und Inhalt des Behältnisses
IMMUNATE ist in folgenden Größen erhältlich: 250 I.E., 500 I.E. und 1000 I.E., aufzulösen in 5 ml (250
I.E. und 500 I.E.) bzw. 10 ml (1000 I.E.) sterilisiertem Wasser für Injektionszwecke. Sowohl Pulver als auch Lösungsmittel sind in Einzeldosis-Glasfläschchen mit Gummi-Schutzkappe abgefüllt. Jede Packung enthält auch ein Set zum Auflösen und Verabreichen.
6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung
IMMUNATE erst unmittelbar vor der Verabreichung auflösen. Die fertige Lösung sofort verwenden (sie enthält keine Konservierungsmittel).
Lösungen, die trüb sind oder Ablagerungen aufweisen, nicht verwenden.
Auflösen des Pulvers: Auf aseptische Arbeitsweise achten.
1. Die ungeöffnete Lösungsmittelflasche auf Raumtemperatur bringen (max. 37 °C).
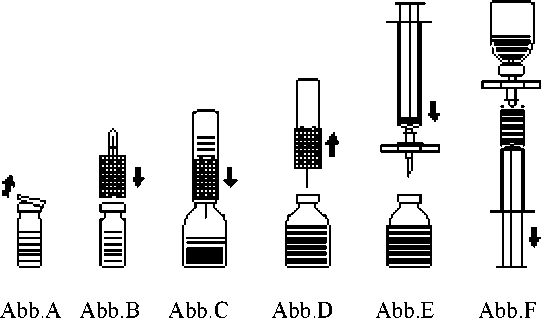
2. Die Schutzkappen von den Flaschen mit Pulver und Lösungsmittel entfernen (Abb. A) und die Gummistopfen beider Flaschen desinfizieren.
3. Transferset mit der gewellten Seite auf die Lösungsmittelflasche aufsetzen und eindrücken (Abb. B).
4. Schutzkappe von der anderen Seite des Transfersets abziehen. Freies Kanülenende nicht berühren!
5. Transferset mit aufgesetzter Wasserflasche von oben in die Flasche mit Pulver einstechen (Abb. C). Durch das in der Flasche mit Pulver bestehende Vakuum wird das Lösungsmittel angesaugt.
6. Nach etwa einer Minute Lösungsmittelflasche samt Transferset von der Konzentratflasche abziehen (Abb. D). Da sich das Präparat rasch löst, die Konzentratflasche, wenn überhaupt, nur vorsichtig schwenken. DEN INHALT DER KONZENTRATFLASCHE NICHT SCHÜTTELN! DIE KONZENTRATFLASCHE ERST UNMITTELBAR VOR DER ENTNAHME DES INHALTS UMDREHEN.
7. Parenterale Arzneimittelprodukte nach dem Auflösen und vor der Verabreichung stets visuell auf Partikel und Verfärbung überprüfen. Gelegentlich können jedoch wenige kleine Partikel sichtbar sein, auch wenn das Auflösen strikt nach Anleitung durchgeführt wurde. Das mitgelieferte Filterset entfernt diese Partikel, ohne die auf der Packung angegebene Konzentration des arzneilich wirksamen Bestandteils zu verringern.
Anwendung: Auf aseptische Arbeitsweise achten
1. Um zu verhindern, dass vom Stopfen ausgestochene Gummipartikel verabreicht werden (Gefahr von Mikroembolien) ist zur Entnahme des gelösten Präparats das beigepackte Filterset zu benutzen. Filterset auf die beigepackte Einmalspritze setzen und in den Gummistopfen einstechen (Abb. E).
2. Durch zwischenzeitliches Abziehen der Spritze vom Filterset wird die Konzentratflasche belüftet, wodurch eventuell entstandener Schaum zusammenfällt. Daraufhin die Injektionslösung durch das Filterset in die Spritze aufziehen (Abb. F).
3. Anschließend Spritze vom Filterset abziehen und die Lösung mit Hilfe des mitgelieferten Infusionssets (oder der mitgelieferten Einmalkanüle) langsam intravenös applizieren (maximale Injektionsrate: 2 ml pro Minute).
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
7. INHABER DER ZULASSUNG
Baxter Deutschland GmbH Edisonstraße 4 85716 Unterschleißheim Tel.: 089/31701-0 Fax: 089/31701-177 E-Mail: info de@baxter.com
8. ZULASSUNGSNUMMERN
3854.01.00
3854.02.00
3854.03.00
9. DATUM DER ERTEILUNG DER ZULASSUNG/VERLÄNGERUNG DER ZULASSUNG
Datum der Zulassung: 20. Oktober 1998
Datum der Verlängerung der Zulassung: 20. Oktober 2003
10. STAND DER INFORMATION
Dezember 2014
11. SONSTIGE HINWEISE
Herkunftsländer der zur Produktion verwendeten Plasmen:
Deutschland, Finnland, Norwegen, Österreich, Schweden, Schweiz, Tschechien und Vereinigte Staaten von Amerika.
Es wird auf die Dokumentationspflicht gemäß Transfusionsgesetz hingewiesen.
Baxter ist eine Marke der Baxter International Inc. IMMUNATE ist eine Marke der Baxter AG.
FI IMMUNATE Seite 13 von 13