Lovadura 20 Mg
Wortlaut der für die Fachinformation vorgesehenen Angaben
Fachinformation
1. Bezeichnung der Arzneimittel
Lovadura 20 mg, Tabletten Lovadura 40 mg, Tabletten
2. Qualitative und quantitative Zusammensetzung Lovadura 20 mg:
1 Tablette enthält 20 mg Lovastatin Lovadura 40 mg:
1 Tablette enthält 40 mg Lovastatin
Die vollständige Auflistung der sonstigen Bestandteile siehe Abschnitt 6.1.
3. Darreichungsform Tablette
Lovadura 20 mg:
hellblaue, achteckige, plane Tablette mit leichter Facette, einseitiger Teilungsrille und Prägungen "LV" u. "20" ober- und unterhalb der T eilungsrille und "G" auf Rückseite Die Tablette kann in gleiche Hälften geteilt werden.
Lovadura 40 mg:
schwach hellgrüne, achteckige, plane Tablette mit leichter Facette, einseitiger Teilungsrille und Prägungen "LV" u. "40" ober- und unterhalb der Teilungsrille und "G" auf Rückseite Die Tablette kann in gleiche Hälften geteilt werden.
4. Klinische Angaben
4.1 Anwendungsgebiete
Zur Senkung erhöhter Gesamt- und LDL-Cholesterinspiegel im Serum, wenn Diät und andere nichtpharmakologische Maßnahmen (z. B. körperliches Training und Gewichtsabnahme) alleine eine ungenügende Wirkung zeigten,
- bei Patienten mit primärer Hypercholesterinämie,
- bei Patienten mit kombinierter Hypercholesterinämie und Hypertriglyceridämie, wenn die Hypercholesterinämie im Vordergrund der therapeutischen Bemühungen steht.
Hinweis
Lovadura besitzt einen nur mäßigen Effekt auf die Triglyceride und ist nicht indiziert, wenn bei der Fettstoffwechselstörung die Hypertriglyceridämie im Vordergrund steht (z. B. Hyperlipidämie der Typen I, IV und V nach Fredrickson).
Für die Hyperlipidämie Typ III liegen keine ausreichenden Erfahrungen vor.
4.2 Dosierung, Art und Dauer der Anwendung
Vor Behandlungsbeginn mit Lovadura sollten geeignete diätetische Maßnahmen zur Senkung erhöhter Cholesterinspiegel ergriffen werden. Das Diätschema sollte auch während der Behandlung mit Lovadura fortgesetzt werden.
Hypercholesterinämie
Die empfohlene Anfangsdosierung beträgt täglich 1 Tablette Lovadura 20 mg (20 mg Lovastatin) oder eine halbe Tablette Lovadura 40 mg (20 mg Lovastatin) mit dem Abendessen.
Patienten mit leichter bis mittelschwerer Hypercholesterinämie können mit einer Anfangsdosierung von 10 mg Lovastatin behandelt werden.
Falls erforderlich sollten Anpassungen der Dosierung anhand der PlasmaCholesterinkonzentrationen bis zu einer Höchstdosis von 80 mg Lovastatin pro Tag in Intervallen von mindestens 4 Wochen erfolgen, wobei Lovastatin als Einzeldosis oder als zwei halbe Dosen zum Frühstück und zum Abendessen eingenommen werden kann.
Die Dosierung sollte reduziert werden bei Absinken des LDL-Cholesterinspiegels unter 75 mg/dl (1,94 mmol/l) oder des Gesamt-Cholesterinspiegels unter 140 mg/dl (3,6 mmol/l).
Koronaratherosklerose
In Studien zur Koronaratherosklerose, in denen Lovastatin mit oder ohne Begleittherapie eingesetzt, wurde, wurden Dosierungen von 20 mg bis 80 mg täglich verwendet, die als Einzeldosis oder in geteilten Dosen verabreicht wurden. In den beiden Studien, in denen Lovastatin allein verwendet wurde, wurde die Dosis reduziert, wenn der GesamtCholesterinspiegel im Plasma auf unter 110 mg/dL (2,85 mmol/l) bzw. das LDL-Cholesterin unter 80 mg/dL (2,1 mmol/l) gesunken war.
Kombinationstherapie
Lovastatin ist allein oder in Kombination mit Gallensäuren-Ionenaustauschern (z. B.
Colestyramin oder Colestipol) wirksam.
Bei gleichzeitiger Gabe von. Ciclosporin, Danazol, Gemfibrozil, anderen Fibraten oder lipidsenkenden Dosen (> 1 g/Tag) von Niacin sollte eine Dosis von 20 mg Lovastatin pro Tag nicht überschritten werden. Bei gleichzeitiger Gabe von Amiodaron oder Verapamil sollte eine Dosis von 40 mg Lovastatin pro Tag nicht überschritten werden (siehe Abschnitt 4.4, Myopathie/Rhabdomyolyse und Abschnitt 4.5).
Dosierung bei Niereninsuffizienz
Bei Patienten mit mäßiger Niereninsuffizienz ist in der Regel keine Dosisanpassung erforderlich, da Lovadura nur in geringem Ausmaß renal eliminiert wird.
Bei Patienten mit erheblich eingeschränkter Nierenfunktion (Kreatinin-Clearance < 30 ml/min), sollte die Entscheidung zur Therapie mit höheren Dosierungen als 20 mg Lovastatin pro Tag sorgfältig abgewogen werden; sofern sie als notwendig erachtet wird, ist sie mit Vorsicht durchzuführen (siehe Abschnitt 4.4, Myopathie/Rhabdomyolyse und Abschnitt 5.1).
Dosierung bei Kindern (10-17 Jahre) mit heterozygoter familiärer Hypercholesterinämie
Der empfohlene Dosierungsbereich liegt bei 10-40 mg/Tag; die empfohlene Höchstdosis beträgt 40 mg pro T ag. Die jeweilige Dosis ist individuell dem jeweiligen empfohlenen Therapieziel anzupassen (siehe Abschnitt 5.1). Patienten, die zum Erreichen ihres Therapieziels eine Senkung des LDL-C von 20 % oder mehr benötigen, sollten mit einer Anfangsdosis von 20 mg Lovastatin/Tag beginnen. Eine Anfangsdosis von 10 mg kann bei Patienten in Betracht gezogen werden, bei denen eine geringere Senkung des LDL-C ausreichend ist.
Art der Anwendung
Lovadura soll unzerkaut und mit etwas Flüssigkeit mit der Mahlzeit eingenommen werden.
In kontrollierten Versuchen war eine zum Abendessen eingenommene Tagesdosis wirksamer als die morgendliche Gabe; die erhöhte Wirkung der abendlichen Dosis wird darauf zurückgeführt, dass die Cholesterinbiosynthese vorwiegend in der Nacht stattfindet.
Dauer der Anwendung
Bei unzureichendem Erfolg einer Diät und anderer nicht pharmakologischer Maßnahmen erfordert eine primäre Hypercholesterinämie in der Regel eine Langzeittherapie mit Lovadura.
Anwendung bei Kindern und Jugendlichen (<18 Jahren)
Die Anwendung von Lovadura bei Kindern wird nicht empfohlen, da keine Studien zu Wirksamkeit und Verträglichkeit vorliegen.
Anwendung bei älteren Patienten
Bei Patienten über 60 Jahren, die in einer kontrollierten klinischen Studie Lovastatin erhielten, schien die Wirksamkeit ähnlich der in der übrigen Patientenpopulation zu sein; es zeigte sich kein offensichtlicher Anstieg in der Häufigkeit der klinisch oder labormäßig erfassten Nebenwirkungen.
Homozygote familiäre Hypercholesterinämie
Bei Patienten mit der seltenen homozygoten Form der familiären Hypercholesterinämie war die Wirksamkeit von Lovadura herabgesetzt, möglicherweise weil bei diesen Patienten funktionsfähige LDL-Rezeptoren fehlen. Bei diesem Patientengut scheint es außerdem häufiger zu einem Anstieg der Serum-T ransaminasen zu kommen.
Diabetes mellitus
Es konnte gezeigt werden, dass Lovastatin bei Patienten mit unkompliziertem, gut eingestelltem Diabetes mellitus (Typ I und II) und einer primären Hypercholesterinämie wirksam ist. Die Reduktion der Lipidspiegel im Plasma war vergleichbar mit der bei Patienten ohne Diabetes mellitus. Der Glukosestoffwechsel wurde nicht negativ beeinflusst.
4.3 Gegenanzeigen
- Überempfindlichkeit gegen Lovastatin oder einen der sonstigen Bestandteile
- Aktive Lebererkrankung oder persistierende Erhöhung der Serum-T ransaminasen unklarer Genese
- Schwangerschaft und Stillzeit (siehe Abschnitte 4.4 und 4.6)
- Gleichzeitige Anwendung von potenten CYP3A4-Inhibitoren (z. B. Itraconazol,
Ketoconazol, HIV-Protease-Inhibitoren, Erythromycin, Clarithromycin, Telithromycin und Nefazodon) (siehe Abschnitt 4.5)
4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung Myopathie/Rhabdomyolyse
Wie andere HMG-CoA-Reduktase-Inhibitoren ruft Lovastatin gelegentlich eine Myopathie hervor, die sich in Muskelschmerzen, -empfindlichkeit oder -schwäche verbunden mit ausgeprägten Erhöhungen der Kreatinkinase (CK) (> das Zehnfache des oberen Normwertes) äußert. Bisweilen manifestiert sich die Myopathie als Rhabdomyolyse mit oder ohne akutes Nierenversagen aufgrund von Myoglobinurie, sehr selten mit tödlichem Ausgang. Das Risiko einer Myopathie ist bei hoher HMG-CoA-Reduktase-Inhibitoraktivität im Plasma erhöht.
• Das Risiko einer Myopathie/Rhabdomyolyse ist bei gleichzeitiger Anwendung von Lovastatin mit den folgenden Arzneimitteln erhöht:
Potente Inhibitoren von CYP3A4, z. B. Itraconazol, Ketoconazol, Erythromycin, Clarithromycin, Telithromycin, HIV-Protease-Inhibitoren, Nefazodon; insbesondere bei
Lipidsenker, die bei alleiniger Gabe eine Myopathie verursachen können: Gemfibrozil, andere Fibrate oder lipidsenkende Dosen (> 1 g/Tag) von Niacin, insbesondere mit hohen Dosen von Lovastatin (siehe unten; Abschnitt 4.5, Wechselwirkungen mit lipidsenkenden Arzneimitteln, die bei Monotherapie eine Myopathie verursachen können).
Andere Arzneimittel:
Ciclosporin oder Danazol, insbesondere bei hohen Dosen von Lovastatin (siehe Abschnitt 4.5, 'andere Arzneimittel-Wechselwirkungen'; Abschnitte 5.1 und 5.2).
Amiodaron oder Verapamil: Das Risiko einer Myopathie/Rhabdomyolyse ist erhöht, wenn Amiodaron oder Verapamil in Kombination mit höheren Dosen eines nahe verwandten HMG-CoA-Reduktase-Hemmers gleichzeitig angewendet werden (siehe Abschnitt 4.5, Andere Arzneimittel-Wechselwirkungen).
Fusidinsäure: Das Risiko einer Myopathie kann durch die gleichzeitige Anwendung von Fusidinsäure und einem nahe verwandten HMG-CoA-Reduktase-Hemmer erhöht werden (siehe Abschnitt 4.4, Myopathie/Rhabdomyolyse, Abschnitte 5.1 und 5.2).
• Wie bei anderen HMG-CoA-Reduktase-Inhibitoren ist das Risiko für eine Myopathie/Rhabdomyolyse dosisabhängig.
In einer klinischen, randomisierten Studie (EXCEL) trat bei sorgfältiger Überwachung der Patienten und bei Ausschluss einiger interagierender Arzneimittel nur ein einziger Fall von Myopathie unter 4.933 Patienten auf, die 20-40 mg Lovastatin pro Tag über 48 Wochen erhielten; 4 Fälle traten unter 1.649 Patienten auf, die 80 mg Lovastatin pro Tag erhielten.
Messungen der Kreatinkinase (CK)
Die Kreatinkinase (CK) sollte nicht nach körperlicher Anstrengung oder bei Vorliegen anderer plausibler Ursachen für eine CK-Erhöhung gemessen werden, da dies eine Interpretation der Werte erschwert. Wenn die Ausgangswerte der CK signifikant erhöht sind (> das Fünffache des oberen Normwertes), sollte die Messung nach 5-7 Tagen wiederholt werden, um die Ergebnisse zu bestätigen.
Vor Beginn der Therapie
Alle Patienten, die auf Lovastatin eingestellt werden oder deren Lovastatin-Dosis erhöht wird, sollten über das Risiko einer Myopathie aufgeklärt und aufgefordert werden, unklare Muskelschmerzen, -empfindlichkeit oder -schwäche umgehend mitzuteilen.
Bei Patienten mit Risikofaktoren für eine Rhabdomyolyse ist Vorsicht angebracht. Um einen Ausgangswert als Referenz festzustellen, sollten in folgenden Situationen vor Behandlungsbeginn Bestimmungen der CK durchgeführt werden:
• ältere Patienten (> 70 Jahre alt)
• Nierenfunktionsstörung
• unbehandelte Hypothyreose
• hereditäre Muskelerkrankungen in der eigenen oder in der Familienanamnese
• muskuläre Symptomatik unter Behandlung mit Statinen oder Fibraten in der Anamnese
• Alkoholmissbrauch.
In solchen Fällen wird eine sorgfältige Nutzen-Risiko-Abwägung der Behandlung empfohlen. Die betroffenen Patienten sollten engmaschig überwacht werden. Bei Patienten, bei denen bereits eine Myopathie unter Behandlung mit Fibraten oder Statinen aufgetreten ist, sollte die Behandlung mit einer anderen Substanz dieser Klasse nur mit Vorsicht begonnen werden. Wenn die CK-Werte signifikant höher als der Ausgangswert sind (> das Fünffache des oberen Normwertes), sollte nicht mit der Therapie begonnen werden.
Im Behandlungsverlauf
Wenn während der Behandlung mit einem Statin Muskelschmerzen, -schwäche oder Krämpfe auftreten, sollten die CK-Werte bestimmt werden. Wenn die CK-Werte ohne körperliche Anstrengung signifikant erhöht sind (> das Fünffache des oberen Normwertes), ist die Therapie
abzusetzen. Sollte die muskuläre Symptomatik schwerwiegend sein und Beeinträchtigungen verursachen, sollte ein Absetzen der Behandlung in Erwägung gezogen werden, auch wenn die CK-Werte weniger als auf das Fünffache des oberen Normwertes erhöht sind. Bei Verdachtsdiagnose einer Myopathie anderer Ursache sollte die Therapie abgesetzt werden.
Wenn die Symptome verschwinden und die CK-Werte auf den Ausgangswert zurückgehen, kann die erneute Behandlung mit diesem Statin oder mit einem alternativen Statin in der jeweils niedrigsten Dosis und bei engmaschiger Überwachung in Erwägung gezogen werden.
Die Therapie mit Lovastatin sollte einige Tage vor geplanten chirurgischen Eingriffen sowie bei Eintritt eines akuten ernsten Krankheitsbildes bzw. Notwendigkeit von chirurgischen Maßnahmen vorübergehend unterbrochen werden.
Daher:
1. Die gleichzeitige Anwendung von Lovastatin mit CYP3A4-Inhibitoren (z. B. Itraconazol, Ketoconazol, Erythromycin, Clarithromycin, Telithromycin, HIV-Protease-Inhibitoren oder Nefazodon) soll vermieden werden. Falls eine Behandlung mit Itraconazol, Ketoconazol, Erythromycin, Clarithromycin oder Telithromycin unabdingbar ist, muss die Therapie mit Lovastatin während der Behandlungsdauer unterbrochen werden. Die gleichzeitige Anwendung mit anderen in therapeutischen Dosen potenten CYP3A4-Inhibitoren sollte vermieden werden, es sei denn, der Nutzen einer kombinierten Behandlung überwiegt das erhöhte Risiko.
2. Bei gleichzeitiger Behandlung mit Ciclosporin, Danazol, Gemfibrozil, anderen Fibraten oder lipidsenkenden Dosen von Niacin (> 1 g/Tag), sollte eine Dosis von 20 mg Lovastatin pro Tag nicht überschritten werden. Die kombinierte Anwendung von Lovastatin und Gemfibrozil sollte vermieden werden, sofern der Nutzen einer weiteren Senkung des Lipidspiegels das erhöhte Risiko dieser Arzneimittelkombination nicht überwiegt. Der Nutzen der Anwendung von Lovastatin bei Patienten die andere Fibrate, Niacin, Ciclosporin oder Danazol erhalten, sollte sorgfältig gegen das Risiko dieser Arzneimittelkombinationen abgewogen werden. Die zusätzliche Gabe von Fibraten oder Niacin zu Lovastatin führt im Allgemeinen nur zu einer geringen zusätzlichen Senkung von LDL-C, aber es kann eine weitere Senkung von Triglyceriden und ein weiterer Anstieg von HDL-C erreicht werden. Kombinationen von Fibraten oder Niacin mit niedrigen Dosen von Lovastatin wurden in kleinen klinischen Studien zur Kurzzeitanwendung und sorgfältiger Überwachung, ohne Auftreten einer Myopathie angewendet.
3. Die Dosis von Lovastatin sollte bei Patienten, die gleichzeitig Amiodaron oder Verapamil erhalten, 40 mg/Tag nicht überschreiten. Die Kombination von Lovastatin in höheren Dosen als 40 mg pro Tag mit Amiodaron oder Verapamil sollte vermieden werden, sofern der klinische Nutzen das erhöhte Risiko einer Myopathie nicht überwiegt.
4. Patienten unter Fusidinsäure und Lovastatin sollten engmaschig überwacht werden. Eine vorübergehende Unterbrechung der Therapie mit Lovastatin kann in Betracht gezogen werden.
5. Alle Patienten, die auf Lovastatin eingestellt werden oder deren Lovastatin-Dosis erhöht wird, sollten über das Risiko einer Myopathie aufgeklärt und aufgefordert werden, unklare Muskelschmerzen, -empfindlichkeit oder -schwäche umgehend zu melden. Die Lovastatin-Therapie sollte sofort abgesetzt werden, wenn eine Myopathie diagnostiziert oder vermutet wird. Das Vorhandensein dieser Symptome und/oder ein CK-Spiegel über dem Zehnfachen des oberen Normwertes zeigt eine Myopathie an. In den meisten Fällen, bei denen die Behandlung sofort beendet wurde, verschwand die muskuläre Symptomatik und die CK-Werte gingen auf den Ausgangswert zurück. Periodische CK-Bestimmungen können bei Patienten zu Beginn der Therapie mit Lovastatin oder bei Dosiserhöhung in Betracht gezogen werden, aber es gibt keine Sicherheit, dass eine solche Überwachung eine Myopathie verhindern kann.
6. Viele der Patienten, die unter der Therapie mit Lovastatin eine Rhabdomyolyse entwickelt haben, hatten Komplikationen in der Krankengeschichte, einschließlich Niereninsuffizienz in der Regel als Folge eines langjährigen Diabetes mellitus. Solche Patienten bedürfen einer engeren Überwachung. Die Therapie mit Lovastatin sollte einige T age vor geplanten chirurgischen Eingriffen sowie bei Eintritt eines akuten ernsten Krankheitsbildes bzw. Notwendigkeit von chirurgischen Maßnahmen vorübergehend unterbrochen werden.
In sehr seltenen Fällen wurde während oder nach der Behandlung mit einigen Statinen über eine immunvermittelte nekrotisierende Myopathie (immune-mediated necrotizing myopathy; IMNM) berichtet. Die klinischen Charakteristika einer IMNM sind persistierende proximale Muskelschwäche und erhöhte Serum-Kreatinkinase-Werte, die trotz Absetzen der Behandlung mit Statinen fortbestehen.
Auswirkungen auf die Leberfunktion
In den ersten klinischen Studien trat bei einigen wenigen Patienten ein ausgeprägter Anstieg der Serum-Transaminasen (über das 3fache des oberen Normalwerts) auf (gewöhnlich 3-12 Monate nach Beginn der Therapie), jedoch ohne Entwicklung einer Gelbsucht oder anderen klinischen Zeichen oder Symptomen. Es gab keine Anzeichen einer Überempfindlichkeitsreaktion. Bei einem dieser Patienten wurde eine Leberbiopsie durchgeführt, die eine leicht fokale Hepatitis zeigte. Einige dieser Patienten wiesen bereits vor der Therapie mit Lovadura abnorme Leberfunktionswerte auf und/oder konsumierten beträchtliche Mengen Alkohol. Bei denjenigen Patienten, bei denen die Behandlung wegen erhöhter T ransaminasenwerte abgebrochen oder unterbrochen wurde (der Patient eingeschlossen, bei dem die Leberbiopsie durchgeführt wurde), fielen die Transaminasenwerte danach langsam auf die Ausgangswerte ab.
In der 48-Wochen-EXCEL-Studie an 8.245 Patienten zeigten sich bei aufeinander folgenden Kontrollen für Serum-Transaminasenanstiege über den 3fachen oberen Normwert folgende Inzidenzen: unter Placebo 0,1 %, bei 20 mg Lovastatin/Tag 0,1 %, bei 40 mg Lovastatin/Tag 0,9 % sowie bei 80 mg Lovastatin/Tag 1,5 % (siehe Abschnitt 5.1).
Leberfunktionstests werden vor Beginn der Therapie bei Patienten mit einer Lebererkrankung in der Anamnese empfohlen sowie wenn klinisch aus anderen Gründen angezeigt. Es wird empfohlen, die Leberfunktion bei allen Patienten vor Verordnung einer Dosis von 40 mg Lovastatin pro Tag oder mehr und im weiteren Verlauf, wenn klinisch angezeigt, zu kontrollieren.
Bei Anstieg der Serum-T ransaminasen über den 3fachen oberen Normwert soll das potenzielle Risiko einer Fortsetzung der Therapie mit Lovadura gegen die zu erwartenden Vorteile abgewogen werden. Die Transaminasenbestimmungen sollten umgehend wiederholt werden; bei Persistenz oder Progredienz der Abweichungen sollte das Medikament abgesetzt werden.
Wie bei anderen Lipidsenkern traten nach Gabe von Lovadura mäßige Erhöhungen der Transaminasenwerte auf (auf weniger als den 3fachen oberen Normwert) (siehe Abschnitt 4.8). Diese Abweichungen traten bald nach Beginn der Therapie mit Lovadura auf, waren gewöhnlich vorübergehend und nicht von körperlichen Symptomen begleitet. Eine Unterbrechung der Therapie war nicht erforderlich.
Bei Patienten, die in erheblichem Maße Alkohol zu sich nehmen oder Lebererkrankungen in der Anamnese haben, ist die Therapie mit Lovadura besonders sorgfältig zu überwachen; eine aktive Lebererkrankung oder persistierende Erhöhung der Serum-Transaminasen unklarer Genese sind eine Kontraindikation zur Anwendung von Lovastatin (siehe Abschnitt 4.3).
Augenuntersuchungen
Ein altersbedingter Anstieg in der Häufigkeit von Linsentrübungen ist auch ohne Medikamenteneinsatz zu erwarten. Langzeitdaten aus klinischen Studien geben keinen Hinweis, dass Lovastatin eine nachteilige Wirkung auf die menschliche Linse besitzt.
Interstitielle Lungenkrankheit
Bei einigen Statinen wurde, besonders bei Langzeittherapie, in Ausnahmefällen eine interstitielle Lungenkrankheit berichtet (siehe Abschnitt 4.8). Die auftretenden Beschwerden können dabei Dyspnoe, unproduktiven Husten und allgemeine Gesundheitsstörungen (Erschöpfung, Gewichtsverlust und Fieber) einschließen. Wenn vermutet wird, dass ein Patient eine interstitielle Lungenkrankheit entwickelt hat, sollte die Statintherapie abgebrochen werden.
Anwendung bei Kindern und Jugendlichen
Die Sicherheit und Wirksamkeit von Lovastatin bei Patienten mit heterozygoter familiärer Hypercholesterinämie im Alter zwischen 10 und 17 Jahren wurden in kontrollierten klinischen Studien von 48 Wochen Dauer bei heranwachsenden Jungen sowie in kontrollierten klinischen Studien von 24 Wochen Dauer bei Mädchen, mindestens 1 Jahr nach der Menarche, untersucht. Die mit Lovastatin behandelten Patienten wiesen ein Nebenwirkungsprofil auf, das im Allgemeinen dem Profil der mit Placebo behandelten Patienten entsprach. Dosierungen über 40 mg wurden an dieser Population nicht untersucht. In diesen limitierten kontrollierten Studien gab es keine Anzeichen für Auswirkungen auf das Größenwachstum oder die sexuelle Entwicklung bei heranwachsenden Jungen, ebenso wenig wurden Veränderungen an der Dauer des Menstruationszyklus bei Mädchen beobachtet (siehe Abschnitte 4.2, 4.8 und 5.1). Heranwachsende Mädchen sollten über geeignete Verhütungsmethoden während der Lovastatin-Therapie aufgeklärt werden (siehe Abschnitte 4.3 und 4.4, 4.6). Lovastatin wurde weder bei Patienten unter 10 Jahren noch bei präpubertären Kindern untersucht.
Ältere Patienten
Bei Patienten über 60 Jahre, die in einer kontrollierten klinischen Studie Lovastatin erhielten, schien die Wirksamkeit ähnlich der in der übrigen Patientenpopulation zu sein; es zeigte sich kein offensichtlicher Anstieg in der Häufigkeit der klinisch oder labormäßig erfassten Nebenwirkungen.
Homozygote familiäre Hypercholesterinämie
Bei Patienten mit der seltenen homozygoten Form der familiären Hypercholesterinämie war die Wirksamkeit von Lovadura herabgesetzt, möglicherweise weil bei diesen Patienten funktionsfähige LDL-Rezeptoren fehlen. Bei diesen homozygoten Patienten scheint Lovastatin außerdem häufiger zu einem Anstieg der Serum-Transaminasen zu führen (siehe Abschnitt 4.8).
Hypertriglyceridämie
Lovadura besitzt einen nur mäßigen Effekt auf die Triglyceride und ist nicht indiziert, wenn bei der Fettstoffwechselstörung die Hypertriglyceridämie im Vordergrund steht (z. B. Hyperlipidämie der Typen I, IV und V).
Diabetes mellitus
Es gibt Hinweise darauf, dass Statine als Substanzklasse den Blutzuckerspiegel erhöhen und bei manchen Patienten, die ein hohes Risiko für die Entwicklung eines zukünftigen Diabetes mellitus haben, eine Hyperglykämie hervorrufen können, der eine adäquate Diabetesbehandlung erfordert. Dieses Risiko wird jedoch von der Reduktion des vaskulären Risikos durch Statine aufgewogen und sollte daher nicht zu einem Abbruch der Statinbehandlung führen. In Übereinstimmung mit nationalen Richtlinien sollten Risikopatienten (Nüchternblutzucker von 5,6 bis 6,9 mmol/L, BMI>30kg/m , erhöhte Triglyzeridwerte, Hypertonie) sowohl klinisch als auch in Bezug auf die relevanten Laborwerte überwacht werden.
4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
C YP3A4-Wechselwirkungen
Lovastatin wird über CYP3A4 metabolisiert, hat aber keinen inhibitorischen Effekt auf CYP3A4. Deshalb ist auch nicht zu erwarten, dass es die Plasmaspiegel anderer Medikamente, die über CYP3A4 metabolisiert werden, beeinflusst. Starke Inhibitoren von CYP3A4 (siehe unten) erhöhen das Risiko für eine Myopathie wegen einer reduzierten Elimination von Lovastatin.
Itraconazol
Ketoconazol
Erythromycin
Clarithromycin
Telithromycin
HIV-Protease-Inhibitoren
Nefazodon
Wechselwirkungen mit lipidsenkenden Arzneimitteln, die bei Monotherapie eine Myopathie verursachen können
Das Risiko einer Myopathie ist auch durch die folgenden Lipidsenker erhöht, die keine potenten Inhibitoren von CYP3A4 sind, die aber bei Monotherapie eine Myopathie verursachen können.
Siehe Abschnitt 4.4, Myopathie/Rhabdomyolyse.
Gemfibrozil Andere Fibrate
Niacin (Nikotinsäure) (> 1 g/Tag)
Andere Arzneimittel-Wechselwirkungen
Ciclosporin oder Danazol: Das Risiko für eine Myopathie/Rhabdomyolyse wird durch die gleichzeitige Anwendung von Ciclosporin oder Danazol, insbesondere mit höheren Lovastatin-Dosen erhöht (siehe Abschnitt 4.4, Myopathie/Rhabdomyolyse, Abschnitte 5.1 und 5.2).
Amiodaron oderVerapamil: Das Risiko einer Myopathie/Rhabdomyolyse ist bei gleichzeitiger Anwendung von Amiodaron oder Verapamil und einem nahe verwandten Arzneimittel aus der Klasse der HMG-CoA-Reduktase-Hemmer in höheren Dosen erhöht (siehe Abschnitt 4.4, Myopathie/Rhabdomyolyse).
Fusidinsäure: Das Risiko einer Myopathie kann durch die gleichzeitige Anwendung von Fusidinsäure und einem nahe verwandten HMG-CoA-Reduktase-Hemmer erhöht werden (siehe Abschnitt 4.4, Myopathie/Rhabdomyolyse, Abschnitte 5.1 und 5.2).
Andere Wechselwirkungen
Grapefruitsaft enthält einen oder mehr Bestandteile, die CYP3A4 hemmen und kann die Plasmaspiegel von Arzneimitteln erhöhen, die durch CYP3A4 metabolisiert werden. Die Wirkung üblicher Mengen (ein 250-ml Glas täglich) ist minimal (34 % Erhöhung des aktiven Plasmaspiegels und damit der HMG-CoA-Reduktase-hemmenden Aktivität gemessen durch die Fläche unter der Konzentrations-Zeit-Kurve) und hat keine klinische Relevanz. Genuss großer Mengen (über 1 Liter pro Tag) bei gleichzeitiger Anwendung von Lovastatin führte zu einem signifikanten Anstieg des aktiven Plasmaspiegels und damit der HMG-CoA-Reduktase-hemmenden Aktivität und sollte vermieden werden (siehe Abschnitt 4.4, Myopathie/Rhabdomyolyse).
Cumarinderivate
Bei gleichzeitiger Gabe von Lovastatin und Antikoagulanzien vom Typ der Cumarinderivate kann die Prothrombinzeit bei einigen Patienten verlängert sein. Bei Patienten, die mit Antikoagulanzien behandelt werden, sollte die Prothrombinzeit vor Therapiebeginn mit Lovadura und danach in kürzeren Abständen bestimmt werden, um signifikante Veränderungen der Prothrombinzeit zu verhindern. Nach Stabilisierung der Werte wird die Bestimmung der Prothrombinzeit anschließend in den Zeitabständen empfohlen, wie sie für Patienten unter Therapie mit Cumarinderivaten üblich sind. Das gleiche Vorgehen wird bei einer eventuell notwendigen Dosisänderung von Lovadura empfohlen. Lovastatin-Therapie wurde nicht mit Blutungen oder Veränderungen der Prothrombinzeit bei Patienten in Verbindung gebracht, die keine Antikoagulanzien einnahmen.
Propranolol
Bei gesunden Probanden wurden keine klinisch signifikanten pharmakokinetischen oder pharmakodynamischen Wechselwirkungen zwischen Propranolol und Einzeldosen von Lovadura beobachtet.
Digoxin
Bei Patienten mit Hypercholesterinämie wurde bei gleichzeitiger Verabreichung von Lovastatin und Digoxin kein Einfluss auf die Digoxinplasmakonzentrationen beobachtet.
Andere Begleitmedikationen
In klinischen Studien, in denen Lovastatin gleichzeitig mit Angiotensin-Converting-Enzym(ACE)-Hemmern, Betablockern, Kalziumantagonisten (außer Verapamil), Diuretika und nicht-steroidalen antiphlogistischen Medikamenten (NSAR) sowie mit Antidiabetika (Glibenclamid, Glipizid, Insulin, Chlorpropamid) verabreicht wurde, gab es keinen Hinweis auf klinisch relevante nachteilige Wechselwirkungen.
4.6 Schwangerschaft und Stillzeit
Schwangerschaft
Lovadura ist während der Schwangerschaft kontraindiziert.
Die Sicherheit bei schwangeren Frauen wurde nicht untersucht. Mit Lovastatin wurden keine kontrollierten klinischen Studien mit schwangeren Frauen durchgeführt. Es liegen seltene Berichte über kongenitale Anomalien nach intrauteriner Exposition mit HMG-CoA-Reduktase-Inhibitoren vor. Eine Analyse bisheriger Erfahrungen mit ca. 200 Frauen, die versehentlich Lovastatin oder einen strukturverwandten HMG-CoA-Reduktase-Inhibitor im ersten Trimenon der Schwangerschaft eingenommen hatten, zeigte kein erhöhtes Risiko für kongenitale Anomalien gegenüber der Gesamtpopulation. Diese Fallzahl war statistisch ausreichend, um eine Risikoerhöhung um das 2,5fache oder mehr im Vergleich zu der für eine Gesamtpopulation erwarteten Häufigkeit ausschließen zu können.
Obwohl es keine Anzeichen dafür gibt, dass die Inzidenz kongenitaler Anomalien bei Kindern, deren Mütter Lovastatin oder einen anderen eng verwandten HMG-CoA-Reduktase-Inhibitor eingenommen hatten, von der in der Gesamtpopulation beobachteten abweicht, kann eine Behandlung der Mutter mit Lovastatin beim Fetus die Spiegel der Mevalonsäure senken, welche als Vorstufe der Cholesterinsynthese eine Rolle spielt. Da Atherosklerose eine chronische Erkrankung ist, sollte eine Unterbrechung lipidsenkender Therapien während einer Schwangerschaft im Allgemeinen kaum Auswirkungen auf das mit der primären Hypercholesterinämie verbundene Langzeitrisiko haben. Lovastatin darf daher nicht von Frauen eingenommen werden, die schwanger sind, eine Schwangerschaft planen oder vermuten schwanger zu sein. Die Behandlung mit Lovastatin muss unterbrochen werden, bis die Schwangerschaft beendet oder definitiv ausgeschlossen ist (siehe Abschnitt 4.3).
Stillzeit
Es ist nicht bekannt, ob Lovastatin oder seine Metaboliten in die menschliche Muttermilch übergehen. Da viele Arzneimittel in die Muttermilch übergehen und aufgrund des Potenzials für schwerwiegende unerwünschte Wirkungen bei Säuglingen darf Lovadura von stillenden Frauen nicht angewendet werden (siehe Abschnitt 4.3).
4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Lovadura hat keine oder zu vernachlässigende Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen. Bei der T eilnahme am Straßenverkehr oder dem Bedienen von Maschinen ist jedoch zu berücksichtigen, dass nach Markteinführung selten über Schwindel berichtet wurde.
4.8 Nebenwirkungen
Lovastatin wird im Allgemeinen gut vertragen; zum größten Teil sind die Nebenwirkungen leicht und von vorübergehender Natur.
In der 48-wöchigen erweiterten klinischen Bewertung von Lovastatin (EXCEL-Studie), die Lovastatin mit Placebo verglich, waren die berichteten Nebenwirkungen ähnlich denen, der ersten klinischen Studien und die Inzidenz bei Wirkstoff und Placebo war statistisch nicht unterschiedlich.
Bei den Häufigkeitsangaben zu Nebenwirkungen werden folgende Kategorien zugrunde gelegt: Sehr häufig (_1/10)
Häufig (_1/100 bis <1/10)
Gelegentlich (_ 1/1.000 bis <1/100)
Selten (_1/10.000 bis <1/1.000)
Sehr selten (<1/10.000)
Nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar)
Die folgenden Häufigkeitsangaben zu Nebenwirkungen beruhen auf Daten von klinischen Studien und Erfahrungen seit der Markteinführung,
Augenerkrankungen:
Häufig: Verschwommensehen
Erkrankungen des Gastrointestinaltrakts:
Häufig: Verstopfung, Dyspepsie, Bauchschmerzen, Durchfall, Blähungen, Übelkeit,
Gelegentlich: Mundtrockenheit
Selten: Erbrechen, Pankreatitis, Stomatitis
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort:
Gelegentlich: Müdigkeit Selten: Ödeme
Funktionsstörungen der Leber:
Selten: cholestatischer Ikterus, Hepatitis
Stoffwechsel- und Ernährungsstörungen:
Selten: Appetitlosigkeit
Skelettmuskulatur- und Bindegewebserkrankungen:
Häufig: Myalgie, Muskelkrämpfe
Selten: Myopathie, Rhabdomyolyse (siehe Abschnitt 4.4)
Häufigkeit nicht bekannt: Immunvermittelte nekrotisierende Myopathie (siehe Abschnitt 4.4)
Erkrankungen des Nervensystems:
Häufig: Schwindel), Kopfschmerzen,
Gelegentlich: Geschmacksstörungen (Veränderungen der Geschmacksempfindung),
Selten: Parästhesien, periphere Neuropathie Nicht bekannt: Gedächtnisverlust
Psychiatrische Erkrankungen:
Gelegentlich: Schlaflosigkeit, Schlafstörungen
Selten: psychische Störungen einschließlich Angstzustände, Depression,
Nicht bekannt: Alpträume
Erkrankungen der Haut und des Unterhautzellgewebes:
Häufig: Hautausschlag Gelegentlich: Juckreiz
Selten: Alopezie, Erythema multiforme einschließlich Stevens-Johnson-Syndrom, toxische epidermale Nekrolyse
Erkrankungen der Geschlechtsorgane und der Brustdrüse:
Selten: erektile Dysfunktion Nicht bekannt: sexuelle Dysfunktion
Erkrankungen des Immunsystems
Selten wurde über ein offensichtliches Hypersensitivitätssyndrom berichtet, das mit einem oder mehreren der folgenden Symptome einherging: Anaphylaxie, Angioödem, lupus-ähnliches Syndrom, Polymyalgia rheumatica, Dermatomyositis, Vaskulitis, Thrombozytopenie, Leukozytopenie, Eosinophilie, hämolytische Anämie, positive antinukleäre Antikörper und Beschleunigung der Blutsenkungsgeschwindigkeit, Arthritis, Arthralgie, Urtikaria, Asthenie, Photosensitivität, Fieber, Gesichtsrötung, Schüttelfrost, Dyspnoe sowie allgemeines Krankheitsgefühl.
Untersuchungen:
Selten: erhöhte Transaminasen (über dem Dreifachen des oberen Normwertes durch wiederholten Test bestätigt; siehe Abschnitt 4.4, Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung, Auswirkungen auf die Leberfunktion)
Selten: Normwertabweichungen anderer Leberfunktionsparameter einschließlich Erhöhung der alkalischen Phosphatase und des Bilirubins; Anstieg der CK-Werte im Serum (nicht kardiale Fraktion der CK). (Siehe Abschnitt 4.4. , Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung, Myopathie/Rhabdomyolyse)
Die folgenden Nebenwirkungen wurden bei einigen Statinen berichtet:
- Schlafstörungen, wie Schlaflosigkeit und Alpträume
- Gedächtnisverlust
- Störung der Sexualfunktion
- Depressionen
- in Ausnahmefällen und besonders bei Langzeittherapie eine interstitielle Lungenkrankheit (siehe Abschnitt 4.4).
- Diabetes mellitus: Die Häufigkeit ist abhängig von dem Vorhandensein oder dem Fehlen von Risikofaktoren (Nüchternblutzucker > 5.6 mmol/L, BMI>30kg/m , erhöhte Triglyzeridwerte, bestehende Hypertonie).
Laborwertuntersuchungen
| Ausgeprägte und anhaltend erhöhte Serum-Transaminasen wurden selten berichtet (siehe
Abschnitt 4.4). Normwertabweichungen anderer Leberfunktionsparameter einschließlich Erhöhung der alkalischen Phosphatase und des Bilirubins wurden berichtet. Anstieg der CK-Werte im Serum wurde berichtet (nicht kardiale Fraktion der CK). Diese waren in der Regel leicht und vorübergehend; deutliche Erhöhungen wurden selten berichtet (siehe Abschnitt 4.4, Myopathie/Rhabdomyolyse).
Kinder und Jugendliche (10 bis 17 Jahre)
In einer 48-wöchigen Studie bei heranwachsenden Jungen mit heterozygoter familiärer Hypercholesterinämie (n=132) und in einer 24-wöchigen Studie bei Mädchen, mindestens 1 Jahr nach der Menarche mit heterozygoter familiärer Hypercholesterinämie (n=54), war das Sicherheits- und Verträglichkeitsprofil der mit Lovastatin (10 bis 40 mg) behandelten Gruppe im Allgemeinen dem Profil der Placebo-Gruppe ähnlich (siehe Abschnitt 4.4, Anwendung bei Kindern und Jugendlichen, Abschnitt 5.1).
4.9 Überdosierung
Bis weitere Erfahrungen vorliegen, kann eine spezifische Behandlung einer Überdosierung mit Lovastatin nicht empfohlen werden. Allgemeine Maßnahmen sind angezeigt und die Leberfunktion sollte überwacht werden.
Zurzeit ist nicht bekannt, ob Lovastatin und seine Metaboliten dialysabel sind.
Fünf gesunde Probanden erhielten bis zu 200 mg Lovastatin als Einzeldosis, ohne dass es zu klinisch signifikanten nachteiligen Begleiterscheinungen kam. Es wurde über einige Fälle mit akzidenteller Überdosierung berichtet; bei keinem der Patienten kam es zu spezifischen Symptomen oder Folgeerscheinungen. Die maximal eingenommene Dosis betrug 5-6 g Lovastatin.
5. Pharmakologische Eigenschaften
5.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Cholesterin-Synthese-Hemmer/HMG-CoA-Reduktase-
Hemmer/Lipidsenker
ATC-Code: C10A A02
Lovastatin ist ein Fermentationsprodukt aus dem Bodenpilz Aspergillus terreus und die inaktive Vorstufe einer hochspezifischen Verbindung (IC50=2,7-11,1 nmol), welche die endogene Cholesterinsynthese hemmt.
Die biologisch aktive Form von Lovastatin entsteht unmittelbar nach der gastrointestinalen Resorption in vivo. Diese inhibiert kompetitiv die 3-Hydroxy-3-methylglutaryl-Coenzym A(HMG-CoA)-Reduktase. Die Aktivität dieses Schlüsselenzyms bestimmt die Bildung von Mevalonsäure und daher das Ausmaß der Neusynthese von Cholesterin. Die Hemmung dieses „Schrittmacher“-Enzyms der Cholesterinsynthese durch Lovastatin führt zu einer Abnahme der intrazellulären Cholesterinkonzentration. Diese signalisiert der Zelle, dass vermehrt Cholesterin, welches im Gefäßsystem hauptsächlich in Form der LDL transportiert wird, über ein spezialisiertes T ransportsystem in der Zellmembran - die LDL-Rezeptoren - von außen in die Zelle transportiert werden muss. Durch Geninduktion kommt es daher zu einer gesteigerten Neusynthese von LDL-Rezeptoren, wodurch die vermehrte Aufnahme von zirkulierenden LdL in die Zelle ermöglicht wird.
In klinischen Studien konnte gezeigt werden, dass durch dieses Wirkprinzip - bei dem ein physiologischer Regelmechanismus ausgenutzt wird - Lovastatin im Serum zu einem Abfall des Gesamtcholesterins sowie des LDL- und VLDL-Cholesterins führt. Es zeigte sich, dass eine Senkung der Lipidspiegel sowohl bei normalen als auch bei erhöhten Ausgangswerten auftritt und die Konzentration von Apolipoprotein B bei der Behandlung mit Lovastatin abnimmt. Da jedes LDL-Partikel nur jeweils ein Molekül Apolipoprotein B enthält und letzteres nur in geringfügigen Mengen in anderen Lipoproteinen vorkommt, kann angenommen werden, dass Lovastatin nicht nur zu einer Erniedrigung des Cholesteringehaltes, sondern auch zu einer mengenmäßigen Abnahme der LDL führt. Die cholesterinsenkende Wirkung von Lovastatin scheint überdies auch auf einer Verminderung der VLDL - der Vorstufe von LDL - zu beruhen. Zudem zeigte sich, dass das HDL-Cholesterin unter der Therapie mit Lovastatin mittelgradig ansteigt. Insgesamt resultieren aus diesen Veränderungen eine Abnahme des Verhältnisses von Gesamt- zu HDL-Cholesterin und LDL- zu HDL-Cholesterin. Die Konzentration der Triglyceride nahm ab.
Die Wirksamkeit der Behandlung der Koronarsklerose mit Lovastatin wurde in drei randomisierten placebokontrollierten klinischen Studien von 2-2,5 Jahren Dauer untersucht. Alle Patienten wiesen eine angiographisch, mittels computergestützter quantitativer Koronarangiographie (QCA) gesicherte Koronarsklerose auf.
In der ersten Studie (Canadian Coronary Atherosclerosis Intervention Trial) wurde die Wirksamkeit von Lovastatin in einer Dosierung von 20-80 mg täglich bei 331 Patienten (Männer und Frauen (18 %) bis 70 Jahre) mit Gesamt-Cholesterinwerten zwischen 220-300 mg/dl (5,70-7,77 mmol/l) untersucht. Lovastatin verlangsamte signifikant die Progression der Atherosklerose und senkte den Anteil der Patienten mit neuen Läsionen.
In der zweiten Studie (Monitored Atherosclerosis Regression Study) wurde die Wirksamkeit von Lovastatin in einer Dosierung von 40 mg 2mal täglich bei 270 Patienten (Männer und Frauen (9 %) bis 67 Jahre) mit Gesamt-Cholesterinwerten zwischen 190-295 mg/dl (4,92-7,64 mmol/l) untersucht. In der Auswertung mittels der quantitativen Koronarangiographie ergab sich bei Betrachtung aller Läsionen hinsichtlich der prozentualen Veränderung des Stenosegrades kein statistischer Unterschied zwischen Verum- und Placebogruppe (primärer Endpunkt). Die Angiogramme wurden auch einer Expertengruppe vorgelegt, die die angiographischen Veränderungen übereinstimmend mittels Global Change Score (sekundärer Endpunkt) beurteilte. Nach dieser Methode verlangsamte Lovastatin signifikant das Fortschreiten der atherosklerotischen Erkrankung insgesamt. Die Anzahl der Patienten, die eine Regression zeigten, war unter Lovastatin doppelt so hoch.
In der dritten Studie (Familial Atherosclerosis Treatment Study) wurde die Wirksamkeit einer Kombinationstherapie mit Lovastatin und Colestipol bei 98 Patienten (Männer bis 62 Jahre) mit einer hinsichtlich vaskulärer Erkrankungen positiven Familienanamnese, einem Apolipoprotein-B-Spiegel von > 125 mg/dl und einem durchschnittlichen GesamtCholesterin von 270 mg/dl (6,99 mmol/l) untersucht. Lovastatin und Colestipol reduzierten signifikant die Anzahl progredienter koronarer Läsionen und führten häufiger zu einer Regression.
Die Wirkung von Lovastatin auf das Fortschreiten der Atherosklerose in den Koronararterien ist durch ähnliche Befunde in einem anderen Gefäßsystem bestätigt worden. In der Asymptomatic Carotid Artery Plaque Study (ACAPS) wurde die Wirksamkeit der Therapie der Atherosklerose der Karotiden mit Lovastatin bei Patienten mit frühen asymptomatischen Karotisläsionen, ohne Anzeichen für eine KHK und einem durchschnittlichen Gesamt-Cholesterin von 235 mg/dl (6,1 mmol/l) mittels B-Mode Ultraschall bewertet. In dieser doppelblinden, kontrollierten klinischen Studie wurden 919 Patienten (Männer und Frauen (48,5 %) bis 79 Jahre) randomisiert vier Studiengruppen zugeteilt und mit Placebo, 10-40 mg Lovastatin/Tag und/oder Warfarin behandelt. Mittels Ultrasonographie wurden die Unterschiede der mittleren maximalen Intima-Media-Dicke der Karotiswand pro Patient zu Studienbeginn und nach drei Jahren in 12 untersuchten Abschnitten bestimmt. Lovastatin in Monotherapie führte verglichen mit Placebo zu einer signifikanten Regression der mittleren maximalen Intima-Media-Dicke (IMT). In der Lovastatin-Gruppe (n=460) kam es im Vergleich zur Placebo-Gruppe (n=459) zu einer signifikanten Reduktion der Anzahl an Patienten mit schwerwiegenden kardiovaskulären Ereignissen (5 vs. 14) sowie zu einer signifikanten Reduktion der Gesamtmortalität (1 vs. 8).
Die aktive Form von Lovastatin inhibiert die enzymatische Umwandlung von HMG-CoA in Mevalonsäure. Diese Wirkung ist spezifisch; eine Hemmung anderer körpereigener Enzyme wurde bisher nicht beobachtet. Da der durch Lovastatin beeinflusste Syntheseschritt eine frühe Stufe der Cholesterin-Biosynthese betrifft, ist die Akkumulation von potenziell toxischen Steroiden nicht zu erwarten. Auch kommt es durch Lovastatin nicht zu einer Akkumulation von HMG-CoA, da dieses umgehend in Acetyl-CoA - das Schlüsselprodukt des Intermediärstoffwechsels und Ausgangspunkt zahlreicher biochemischer Reaktionen - zurückverwandelt wird.
Die Umwandlung von HMG-CoA in Mevalonsäure wird durch therapeutische Dosen von Lovastatin nicht vollständig verhindert, so dass genügend Mevalonsäure für körpereigene Synthesezwecke zur Verfügung steht. So konnte gezeigt werden, dass z. B. die Steroidgenese, die auf Mevalonsäure angewiesen ist, nicht beeinträchtigt wird.
Die Lithogenität der Galle wurde durch Lovastatin nicht erhöht; eine steigende Inzidenz von Gallensteinen ist nicht zu erwarten.
Das Ansprechen auf optische Reize, Nervenreizleitungsmessungen und Elektromyographie bei über 30 Patienten, die Lovastatin erhielten, zeigte keine Anzeichen einer neurotoxischen Wirkung.
5.2 Pharmakokinetische Eigenschaften
Lovastatin ist ein Lakton, welches in vivo in der Leber umgehend hydrolysiert wird, wobei die korrespondierende Beta-Hydroxysäure entsteht. Diese ist ein hochspezifischer (IC50 = 2,7-11,1 nmol), kompetitiver Inhibitor der HMG-CoA-Reduktase. Die Hemmung der HMG-CoA-Reduktase ist die Grundlage eines Testsystems zur pharmakokinetischen Bestimmung der Beta-Hydroxysäuremetaboliten (aktive Inhibitoren) und - nach basischer Hydrolyse der Proben - der Gesamtinhibitoren (der Summe aus aktiven und latenten Inhibitoren) im Plasma nach Verabreichung von Lovastatin.
Nach einer oralen Gabe von 14C-markiertem Lovastatin wurden beim Menschen 10 % der Dosis mit dem Harn und 83 % über die Fäzes ausgeschieden. Die 83 %, die mit den Fäzes
ausgeschieden werden, repräsentieren sowohl die Substanzäquivalente, die in die Galle ausgeschieden wurden, als auch die Äquivalente nicht resorbierter Substanz.
Die Resorptionsrate einer oralen Gabe von Lovastatin wurde in vier verschiedenen Tierspezies untersucht, wobei eine intravenöse Dosis als Referenzkriterium herangezogen wurde; sie beträgt nach diesen Versuchen im Mittel etwa 30 %. Studien am Hund haben gezeigt, dass die Verfügbarkeit der resorbierten Substanz im großen Kreislauf durch einen ausgeprägten First-Pass-Effekt in der Leber, dem primären Wirkort, limitiert wird, wobei Substanzäquivalente anschließend in die Galle sezerniert werden. In Tierstudien konnte die hohe Leberselektivität von Lovastatin gezeigt werden. Nach oraler Gabe von Lovastatin wurden in der Leber - dem Zielorgan - wesentlich höhere Konzentrationen als in den peripheren Geweben gemessen.
Sowohl Lovastatin als auch sein Beta-Hydroxysäuremetabolit sind an menschliche Plasmaproteine gebunden (> 95 %). In Tierexperimenten konnte gezeigt werden, dass Lovastatin die Bluthirnschranke und die Plazentaschranke passiert. Nach Gabe von Lovastatin wurden beim Menschen folgende aktive Hauptmetaboliten im Plasma gefunden: das Beta-Hydroxysäurederivat, das 6-Hydroxy-Derivat, das 6-Hydroxy-methyl- und das 6-Exomethylenderivat. Die maximalen Plasmakonzentrationen von sowohl aktiven wie auch Gesamtinhibitoren wurden innerhalb von 2-4 Stunden nach Verabreichung der Dosis erreicht. Die Inhibitorkonzentrationen im Plasma zeigten eine lineare Dosisabhängigkeit bis zu einer Dosis von 120 mg Lovastatin. Bei einem einmal täglichen Verabreichungsmodus wurden Steady-State-Plasmakonzentrationen der Gesamtinhibitoren zwischen dem 2. und 3. T ag nach Therapiebeginn erreicht; sie waren im Durchschnitt 1 ^-mal höher als nach einer Einzeldosis. Wurde Lovastatin auf nüchternen Magen eingenommen, betrugen die Plasmakonzentrationen der aktiven Inhibitoren und Gesamtinhibitoren im Mittel etwa 2/3 derjenigen, die gemessen wurden, wenn Lovastatin unmittelbar im Anschluss an eine standardisierte T estmahlzeit verabreicht wurde.
In einer klinischen Studie zeigte sich bei Patienten mit schwerer Niereninsuffizienz (Kreatinin-Clearance 10-30 ml/min), dass nach Einmalgabe von Lovastatin die Plasmaspiegel der Gesamtinhibitoren etwa zweimal höher waren als bei gesunden Probanden.
Das Risiko für eine Myopathie wird durch hohe HMG-CoA-Reduktase-Inhibitoraktivität im Plasma erhöht. Starke CYP3A4-Inhibitoren können die Plasmaspiegel und die Aktivität der HMG-CoA-Reduktase-Hemmung erhöhen und damit das Risiko für eine Myopathie erhöhen (siehe Abschnitte 4.3, 4.4 und 4.5).
Bioverfügbarkeit
Bei vier Patienten mit Hypercholesterinämie, die eine orale Dosis von Lovastatin erhielten, wurde gemessen, dass weniger als 5 % der verabreichten Dosis den großen Kreislauf in Form von aktiven Inhibitoren erreichen.
5.3 Präklinische Daten zur Sicherheit
Die wiederholte Verabreichung hoher Dosen von Lovastatin führte bei verschiedenen Tierspezies zu toxischen Effekten, die auf eine übersteigerte pharmakologische Wirkung zurückzuführen sind, Zielorgane waren vor allem die Leber und das ZNS. In Studien am Hund traten im hohen Dosisbereich nach Gabe von Lovastatin vereinzelt Katarakte auf; auf Basis der Serumspiegel scheint jedoch ein ausreichend hoher Sicherheitsabstand zur humantherapeutischen Dosis zu bestehen.
In einer Batterie von Studien zur genetischen Toxikologie (in-vitro und in-vivo) ergab sich kein Hinweis auf ein genotoxisches Potential.
In Langzeitstudien an Maus und Ratte zur Erfassung eines tumorigenen Potentials wurden nach Gabe von Lovastatin erhöhte Tumorinzidenzen beobachtet:
|
Spezies |
relative Exposition |
beobachtete T umoren |
|
(im Vergleich zur |
|
humantherapeutischen) auf Basis von AUC-Leveln | ||
|
Ratte |
2-7 |
hepatozelluläre Karzinome |
|
Maus |
1-2 |
Papillome im squamösen (nicht-glandulären) Epithel der Magenschleimhaut* |
|
Maus |
3-4 |
hepatozelluläre Karzinome und Adenome |
|
Maus |
4 |
pulmonale Adenome |
* beim Menschen besteht die Magenschleimhaut ausschließlich aus glandulärem Epithel
Die Bedeutung diese Befunde für die Langzeittherapie beim Menschen ist ungeklärt.
In Studien zur Reproduktionstoxikologie verursachte Lovastatin nur nach Verabreichung matern toxischer Dosen Skelettmissbildungen. Zusätzliche Studien zeigten, dass diese wahrscheinlich als Sekundäreffekt materner Toxizität entstanden und nicht auf eine direkte Wirkung von Lovastatin auf die Entwicklung des Feten zurückzuführen sind. Diese Studien zeigten auch, dass Lovastatin die fetalen Mevalonatspiegel senkte, ohne dass ein direkter Nachweis für toxische Wirkungen auf die Feten beobachtet wurde.
Beim Kaninchen wurden bei Dosierungen von bis zu 15 mg/kg/Tag (MTD) keine Missbildungen bei den Nachkommen beobachtet. Testikuläre Degeneration trat beim Hund in Dosierungen ab 20 mg/kg/Tag auf, eine Fertilitätsstudie an der Ratte verlief hingegen negativ.
6. Pharmazeutische Angaben 6.1. Liste der sonstigen Bestandteile Lovadura -20 mg/ -40 mg:
Vorverkleisterte Stärke (Mais); Mikrokristalline Cellulose; Butylhydroxyanisol; Magnesiumstearat (Ph. Eur.).
Lovadura 20 mg zusätzlich:
Indigocarmin, Aluminiumsalz (E 132).
Lovadura 40 mg zusätzlich:
Indigocarmin, Aluminiumsalz (E 132); Chinolingelb, Aluminiumsalz (E 104).
6.2 Inkompatibilitäten Nicht zutreffend.
6.3 Dauer der Haltbarkeit
Die Dauer der Haltbarkeit von Lovadura 10 mg, Lovadura 20 mg und Lovadura 40 mg beträgt 3 Jahre.
6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Für dieses Arzneimittel sind keine besonderen Lagerbedingungen erforderlich
6.5 Art und Inhalt des Behältnisses Aluminium-Al/OPA/PVC-Blister Packungen mit 20, 30, 50 und 100 Tabletten
6.6 Besondere Hinweise für die Beseitigung und sonstige Hinweise zur Handhabung Keine besonderen Anforderungen.
Inhaber der Zulassung
7.
Mylan dura GmbH Postfach 10 06 35 64206 Darmstadt
8. Zulassungsnummern
Lovadura 20 mg:
49036.01.00
Lovadura 40 mg:
49036.02.00
9. Datum der Zulassung / Verlängerung der Zulassung 20.01.2003 / 07.09.2009
10. Stand der Information Februar 2015
11. Verkaufsabgrenzung Verschreibungspflichtig
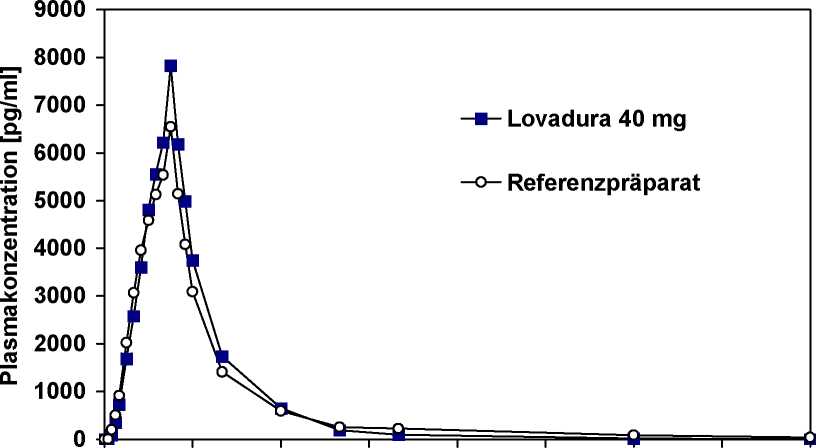
Eine im Jahr 1999 durchgeführte vergleichende Bioverfügbarkeitsuntersuchung (offen, crossover, randomisiert) an 36 gesunden männlichen Probanden (18-53 Jahre) ergab nach Einmalgabe von 40 mg (Dosierung jeweils nach standardisierter Mahlzeit):
Lovastatin
maximale Plasmakonzentration (cmax) in pg/ml:
Zeitpunkt der maximalen Plasmakonzentration (tmax) in h:
Fläche unter der Konzentrations-Zeit-Kurve (AUC0-¥) in pg x h/ml:
|
Lovadura 40 mg |
Referenzpräparat |
|
6444 ± 3484 |
6488 ± 4064 |
|
2,17 ± 1,02 |
1,70 ± 0,70 |
|
24813±12237 |
27493±16272 |
Angabe der Werte als arithmetisches Mittel und Standardabweichung
Mittlere Plasmaspiegelverläufe im Vergleich zu einem Referenzpräparat in einem KonzentrationsZeit-Diagramm:
0 4 8 12 16 20 24
Zeit [h]
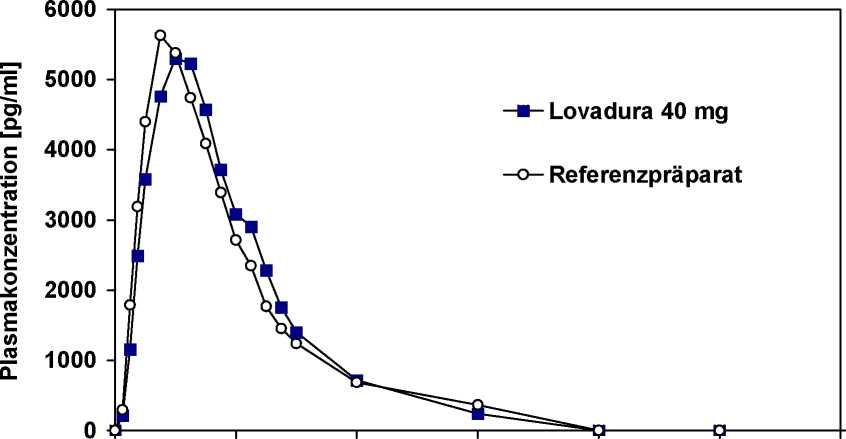
Für das ß-Hydroxysäurederivat (aktiver Metabolit) im Vergleich zum Referenzpräparat:
maximale Plasmakonzentration (cmax) [pg/ml]
Zeitpunkt der maximalen Plasmakonzentration (tmax) [h]
Fläche unter der Konzentrations-ZeitKurve
(AUC0-») [pg x h/ml]
Lovadura 40 mg 8149±4257 4,17 ± 0,68
37514±18147
Referenzpräparat 6905 ± 3560 4,13 ± 0,86
33925±16885
Angabe der Werte als arithmetisches Mittel und Standardabweichung
Mittlere Plasmaspiegelverläufe im Vergleich zu einem Referenzpräparat in einem KonzentrationsZeit-Diagramm: